
Title: Determination of Dissolution Test (IP)
1. Objective: Determination of Dissolution test for tablets and Capsules as per IP Standard.
2. Principle: Dissolution test is done to verify the release of drug in the solution from the tablet because binders, granulation, mixing and coating may affect the release of drug from tablets.
3. Procedure:
3.1 Dissolution test :
The amount of dissolved active ingredient is known as D in dissolution test. The limit of D may be different in different monographs according to the nature of the formulation and its active ingredients. Dissolution test is done using 6 units or dosage forms. These dosages forms are run for the specified time period, sampled and analyzed for the dissolved amount of active ingredient in percentage. This is first stage of the dissolution and known as S1 Stage.
In S1 stage dissolved amount of each unit should not be less than D+5%. It shows that every unit should be above the 5% of the specified limit in the individual monograph.
If any of the unit is found below this limit then we have to analyze the sample in S2 stage.
Six additional units are tested for the dissolved content. The average of the all 12 units should not be less than D and no unit should be less than D-15%. Average should be equal to or more then D but some units may below the D. If any unit is found below the D-15 or average of all units is less than D then sample is analyzed in S3 stage.
Twelve more units are analyzed for the dissolved active content. At this stage the average of all 24 units should not be less than D, only two units may below the D-15% and no unit should be less than Q-25%. This stage gives more flexibility to the sample because average of 24 units should be equal to or more then D but two units may up to D-25
Dissolution stages give the flexibility to the sample that is unable to pass the dissolution test. These stages are accepted by all regulatory bodies. Hence, it is a widely accepted test method for the dissolution of solid dosage form
Use Apparatus I unless otherwise directed. All parts of the apparatus that may come into contact with the preparation under examination or with the dissolution medium are chemically inert and do not adsorb, react or interfere with the preparation under examination. All metal parts of the apparatus that may come into contact with the preparation or the dissolution medium must be made from stainless steel, type 316 or equivalent or coated with a suitable material to ensure that such parts do not react or interfere with the preparation under examination or the dissolution medium. No part of the assembly, including the environment in which the assembly is placed, contributes significant motion, agitation or vibration beyond that due to the smoothly rotating element.
An apparatus that permits observation of the preparation under examination and the stirrer during the test is preferable.
Apparatus -1
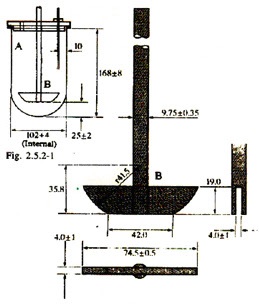
An assembly consisting of the following:
- A cylindrical vessel, A, made of borosilicate glass or any other suitable transparent material, with a hemispherical bottom and with a nominal capacity of 1000 ml and an inside diameter of 98-106 mm (Fig.). The vessel has a flanged upper rim and is fitted with a lid that has a number of openings, one of which is central.
- A motor with a speed regulator capable of maintaining the speed of rotation of the paddle within 4 per cent of that specified in the individual monograph. The motor is fitted with a stirring element which consists of a drive shaft and blade forming a paddle, B (Fig.).
The blade passes through the diameter of the shaft so that the bottom of the blade is flush with the bottom of the shaft. The shaft is positioned so that its axis is within 2 mm of the axis of the vessel and the lower edge of the blade is 23 to 27 mm from the inside bottom of the vessel. The apparatus operates in such a way that the paddle rotates smoothly and without significant wobble.
3. A water-bath set to maintain the dissolution medium at 36.5°C to 37.5°C. The bath liquid is kept in constant and smooth motion during the test. The vessel is securely clamped in the water bath in such a way that the displacement vibration from other equipment, including the water circulation device, is minimized
Apparatus -2
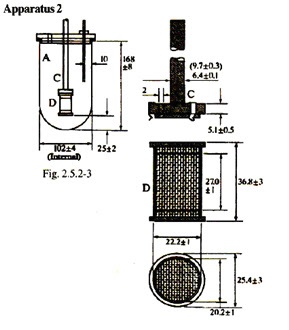
The assembly is the same as in Apparatus 1 except that in the stirring element the paddle is replaced by a basket, D (see Figs.). The metallic shaft rotates smoothly and without significant wobble. The basket consists of two components. The top part, with a vent, is attached to the shaft C, it is fitted with three spring clips, or other suitable means, that allow removal of the lower part for introduction of the preparation under examination and that firmly hold the lower part of the basket concentric with the axis of the vessel during rotation. The lower detachable part of the basket is made of welded-steam cloth, with a wire thickness of 0.254 mm diameter and with 0.381 mm square openings, formed into a cylinder with narrow rim of sheet metal around the top and the bottom. The basket may be plated with a 2.5 mm layer of gold for use with acidic media. The distance between the inside bottom of the vessel and the basket is maintained at 23 to 27 mm during the test.
Dissolution medium
Use the dissolution medium specified in the individual monograph. If the medium is a buffered solution, adjust the solution so that its pH is within 0.05 units of the pH specified in the monograph. The dissolution medium should be deaerated prior to testing.
Time
Where a single time specification is given in the monograph, the test may be concluded in a shorter period if the requirement for the minimum amount dissolved is met. If two or more times are specified, specimen are to be withdrawn only at the stated times, within a tolerance of ± 2 per cent.
Method
Conventional and prolonged-release solid dosage forms
Place the stated volume of the dissolution medium, free from dissolved air, into the vessel of the apparatus. Assemble the apparatus and warm the dissolution medium to 36.5°C to 37.5°C. Unless otherwise stated, place one dosage unit in the apparatus, taking care to exclude air bubbles from the surface of the dosage unit. When Apparatus 1 is used, allow the tablet or capsule to sink to the bottom of the vessel prior to the rotation of the paddle. A suitable device such as a wire of glass helix may be used to keep horizontal at the bottom of the vessel tablets or capsules that would otherwise float. When Apparatus 2 is used, place the tablet or capsule in a dry basket at the beginning of each test. Lower the basket into position before rotation.
Operate the apparatus immediately at the speed of rotation specified in the individual monograph. Within the time interval specified, or at each of the times stated, withdraw a specimen from a zone midway between the surface of the dissolution medium and the top of the rotating blade or basket, not less than 10 mm from the wall of the vessel. Except in the case of single sampling, add a volume of dissolution medium equal to the volume of the samples withdrawn. Filter the sample solution promptly through a membrane filter disc with an average pore diameter not greater than 1.0 micron. Discard the first few ml of the filtrate. Perform the analysis as directed in the individual monograph. Repeat the whole operation five times. Where two or more tablets or capsules are directed to be placed together in the apparatus, carry out six replicate tests.
For each of the tablet or capsule tested, calculate the amount of dissolved active ingredient in solution as a percentage of the stated amount where two or more tablets or capsules are placed together, determine for each test the amount of active ingredient in solution per tablet or capsules and calculate as a percentage of the stated amount.
Acceptance criteria
Conventional-release dosage forms
Unless otherwise specified, the requirements are met if the quantities of active substance dissolved from the dosage units conform to Table 1. If the results do not conform to the requirements at stage S) given in the table, continue testing with additional dosage units through stages S2 and S3 unless the results conform at stage S2′
Where capsule shells interfere with the analysis, remove the contents of not less than 6 capsules as completely as possible, and dissolve the empty capsule shells in the specified volume of the dissolution medium. Perform the analysis as directed in the individual monograph. Make any necessary correction.
Correction factors should not be greater than 25 per cent of the stated amount
Table 1
| Level | Samples tested | Acceptance criteria |
| S1 | 6 | Each value is not less than D + 5% |
| S2 | 6 | Average value of the 12 dosage units (S1 + S2) is equal to or greater than D and no unit is less than D-15%. |
| S3 | 12 | Average value of 24 dosage units (S1 + S2 + S3) is equal to or greater than D; not more than 2 units are less than D – 15%; no unit is less than D – 25%. |
D = is the amount of dissolved active ingredient specified in the individual monograph, expressed as percentage of label amount.
Prolonged-release dosage forms
Unless otherwise specified, the requirements are met if the quantities of active substance dissolved from the dosage units conform to Table 2. If the results do not conform to the requirements at stage L1 given in the table, continue testing with additional dosage units through stages L2 and L1 unless the results conform at stage L2. The limits embrace each value of D, the amount dissolved at each specified dosing interval. Where more than one range is specified, the acceptance criteria apply to each range.
Table 2
| Level | Samples tested | Acceptance criteria |
| L1 | 6 | No individual value lies outside each of the stated ranges and no individual value is less than the stated amount at the final test time. |
| L2 | 6 | The average value of the 12 dosage units (L1 + L2) lies within each of the stated ranges and is not less than the stated amount at the final test time; none is more than 10% of the labelled content outside each of the stated ranges; and none is more than 10% of labelled content below the stated amount at the final test time. |
| L3 | 12 | The average value of the 24 dosage units (L1 + L2 + L3) lies within the stated ranges and is not less than the stated amount at the final test time; not more than 2 of the 24 dosage units are more than 10% of labelled content outside each of the stated ranges; not more than 2 of the 24 dosage units are more than 10% of labelled content below the stated amount at the final test time; and none of the 24 dosage units is more than 20% of labelled content below the stated content at the final test time; none of the units are more than 20% of labelled content outside each of the stated ranges or more than 20% of labelled content below the stated amount at the final test time |
Modified-release dosage forms. Use method A or Method B.
Method A
Acid stage. Place 750 ml of a.1M hydrochloric acid in the vessel, and assemble the apparatus. Warm the dissolution medium to 36.5°C to 37.5°C. Place one dosage unit in the apparatus, cover the vessel and operate the apparatus at the specified rate. After 2 hours of operation in the acid medium, withdraw an aliquot of the liquid and proceed immediately as directed under Buffer stage. Perform the analysis of the aliquot using a suitable assay method.
Buffer stage
Complete the operations of adding the buffer and adjusting the pH within 5 minutes. With the apparatus operating at the rate specified, add to the medium in the vessel 250 ml of a 0.2 M solution of trisodium phosphate dodecahydrate that has been warmed to 36°C to 37°C. Adjust, if necessary, with 2M hydrochloric acid or 2 M sodium hydroxide to a pH of 6.8 ± 0.05. 2 M hydrochloric acid or 2 M sodium hydroxide to a pH of 6.8 ± 0.05.
Method B
Acid stage. Place 1000 ml of 0.1M hydrochloric acid in the vessel and assemble the apparatus. Warm the dissolution medium to 36°C to 37°C. Place one dosage unit in the apparatus, cover the vessel and operate the apparatus at the specified rate. After 2 hours of operation in the acid medium, withdraw an aliquot of the liquid and proceed immediately as directed under Buffer stage. Perform the analysis of the aliquot using a suitable assay method.
Buffer stage
Use buffer that has previously been warmed to 36°Cto 37°C. Drain the acid from the vessel and add 1000 ml of pH 6.8 phosphate buffer, prepared by mixing 3 volumes of 0.1M hydrochloric acid with 1 volume of 0.2 M solution of trisodium phosphate dodecahydrate and adjusting, if necessary, with 2M hydrochloric acid or 2M sodium hydroxide to a pH of 6.8 ± 0.05. This may also be done by removing from the apparatus the vessel containing the acid and replacing it with another vessel containing the buffer and transferring the dosage unit to the vessel containing the buffer. Continue to operate the apparatus for 45 minutes, or for the specified time. At the end of this period, withdraw an aliquot of the liquid and perform the analysis using a suitable assay method.
Acceptance criteria
Acid stage
Unless otherwise specified, the requirements of this part of the test are met if the quantities, based on the percentage of the labelled content of active substance dissolved from the units tested conform to Table 3. Continue the testing through the 3 levels unless the results of both acid and buffer stages conform at an earlier level.
Table 3
| Level | Samples tested | Acceptance criteria |
| A1 | 6 | No individual value exceeds 10% dissolved |
| A2 | 6 | Average value of the 12 dosage units (A1 + A2) is not more than 10% dissolved, and no individual value is greater than 25% dissolved |
| A3 | 12 | Average value of 24 dosage units (A1 + A2 + A3) is not more than 10% dissolved, and no individual value is greater than 25% dissolved. |
Buffer stage
Unless otherwise specified, the requirements of this part of the test are met if the quantities, based on the percentage of the labelled content of active substance dissolved from the units tested conform to Table 4. Continue the testing through the 3 levels unless the results of both acid and buffer stages conform at an earlier level. The value of D in Table 4 is 75 per cent dissolved unless otherwise specified. The quantity, D, is the specified total amount of active substance dissolved in both the acid and buffer stages, expressed as a percentage of the labelled content
Table 4
| Level | Samples tested | Acceptance criteria |
| B1 | 6 | No value is less than D + 5% |
| B2 | 6 | Average value of the 12 dosage units (B1 + B2) is equal to or greater than D, and no unit is less than D – 15% |
| B3 | 12 | Average value of the 24 dosage units (B1 + B2 + B3) is equal to or greater than D; not more than 2 units are less than D – 15%, and no unit is less than D – 25%. |