Role of Placebo in Formulations
placebo, excipients, Tablets, capsules, Liquid orlas, creams, ointments etc.


Pharma Definition/Abbreviation, Pharma Beginners
placebo, excipients, Tablets, capsules, Liquid orlas, creams, ointments etc.
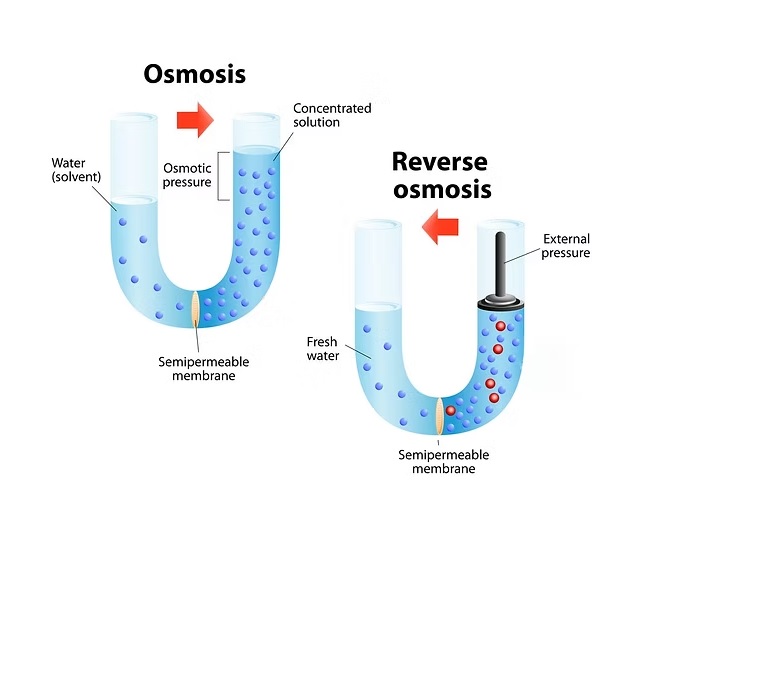
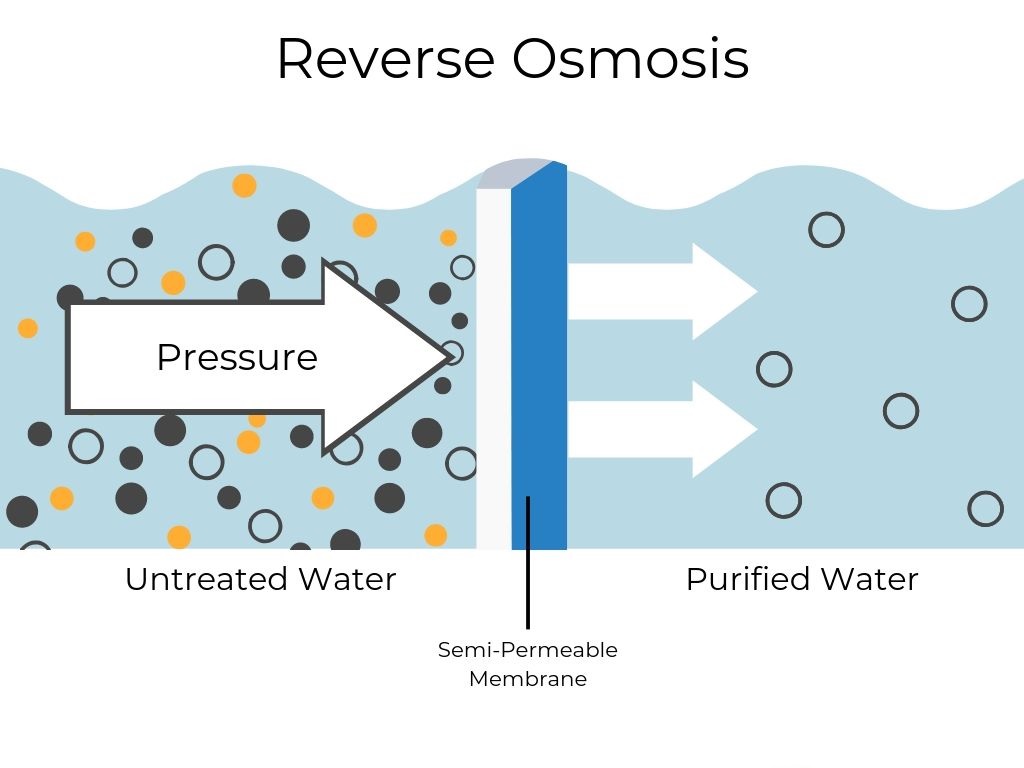
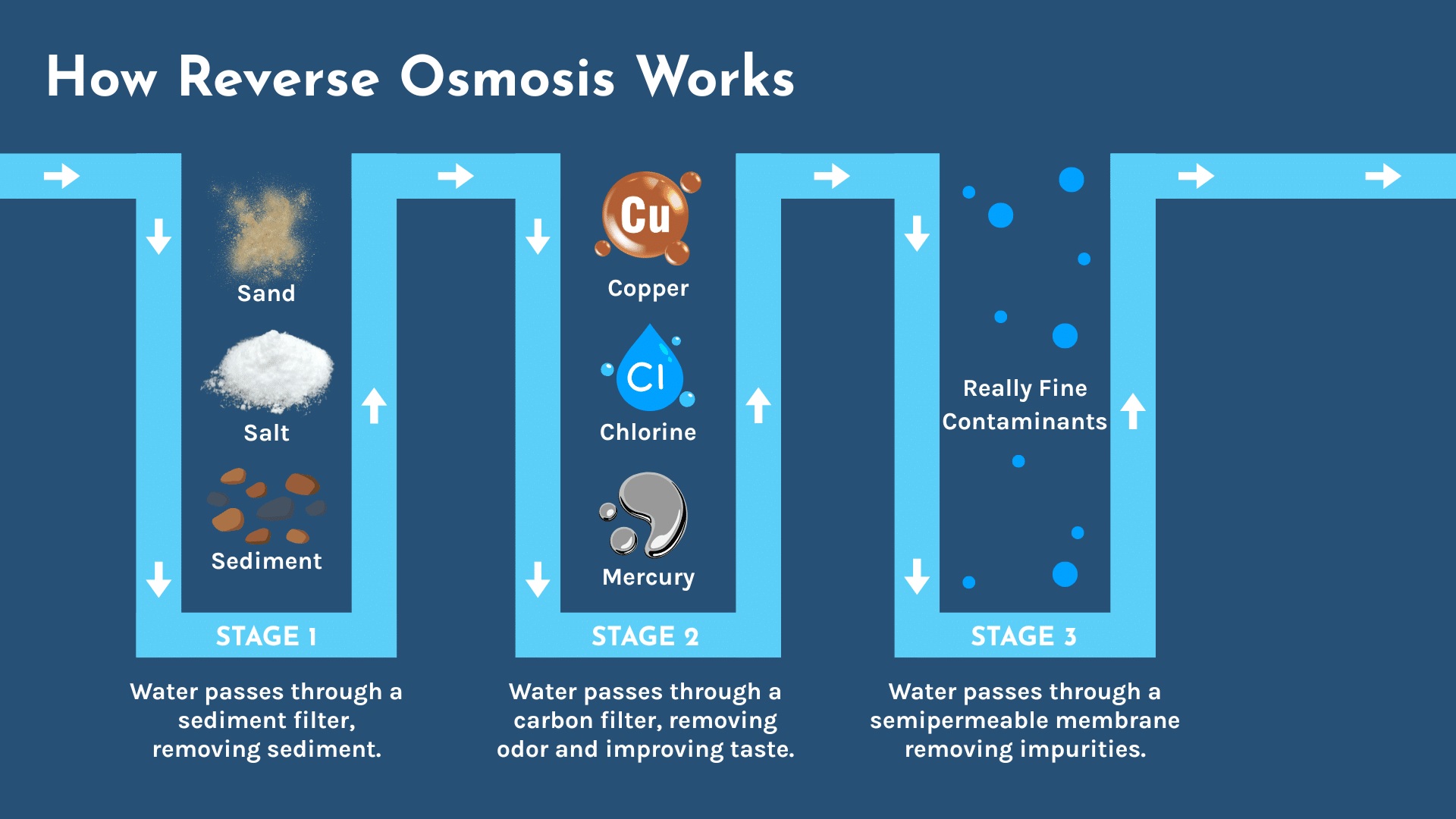
Reverse osmosis (RO) is a water purification method that uses pressure to force water through a semi-permeable membrane, removing contaminants like salt, bacteria, and minerals, resulting in highly purified water for drinking, desalination, and industrial uses. It works by reversing osmosis (the natural movement of water from low to high solute concentration) to move water from a concentrated solution to a purer one, leaving impurities behind.


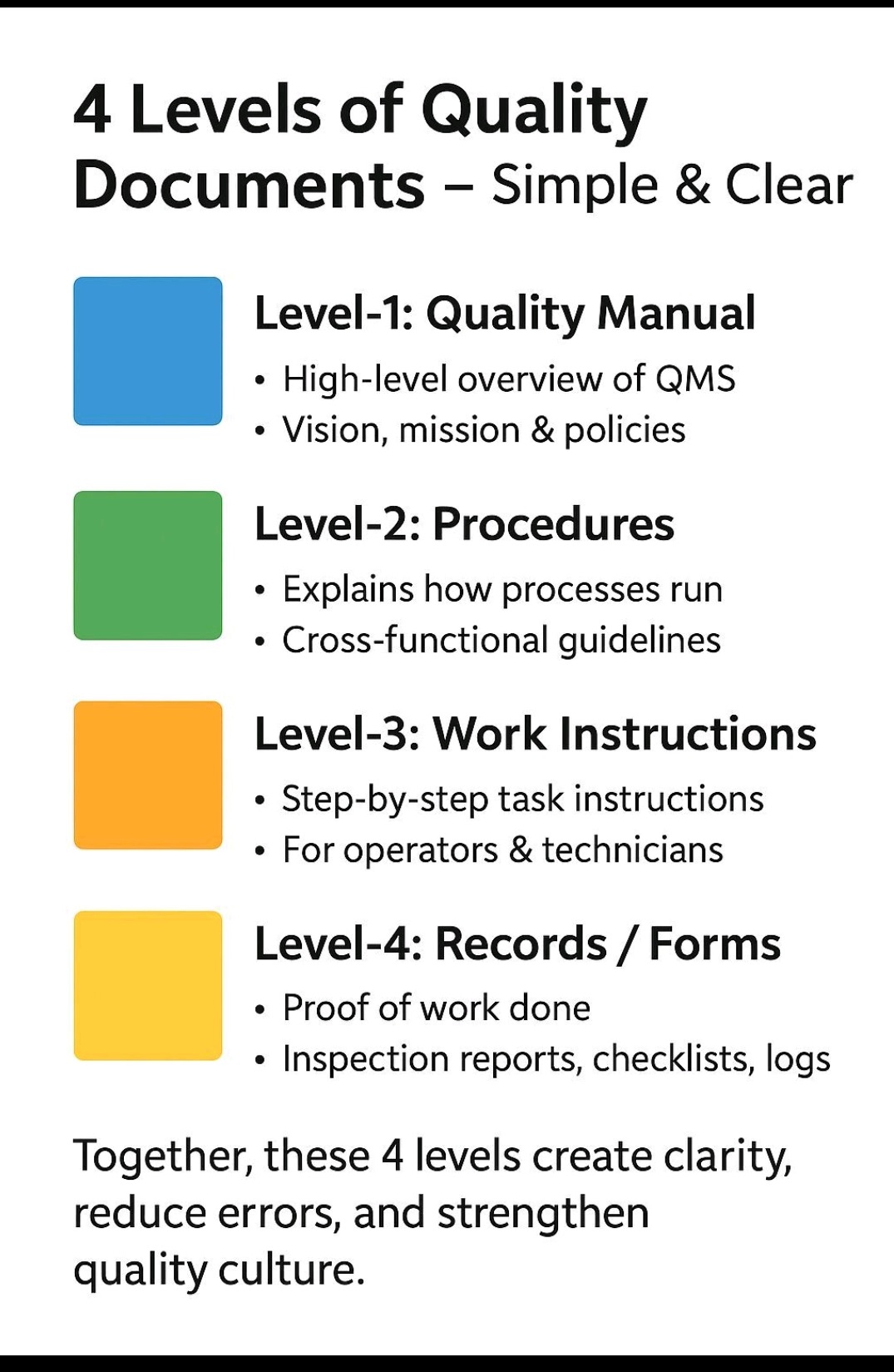
Quality Manual, Quality Policy, Procedures, SOP,s, Work Instructions, Formats, Records, Training,CAPA, Audit, Risk management, Complaint handling, Incident, change control, deviation, calibration, qualification, validation, cGMP,.

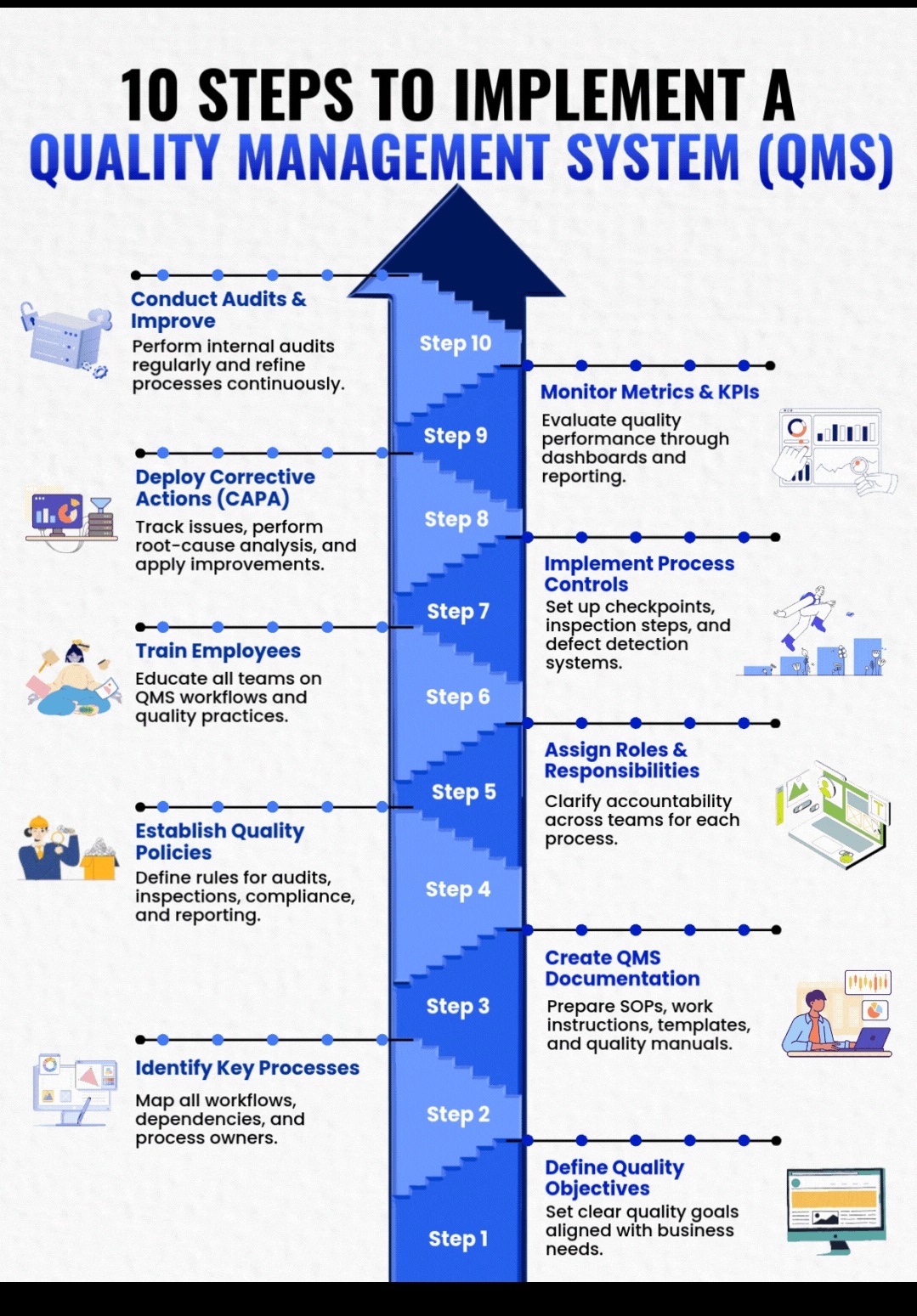

Analytical method validation (AMV) is the documented process of proving an analytical test method is reliable, accurate, and suitable for its specific purpose.
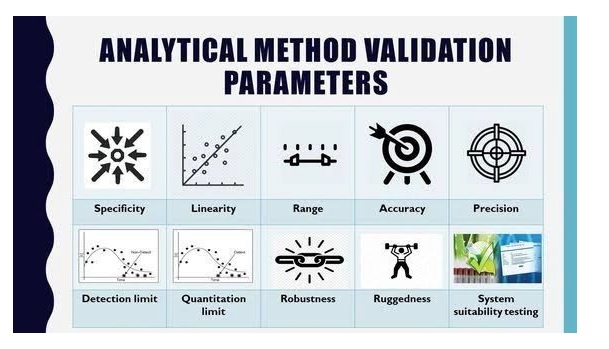

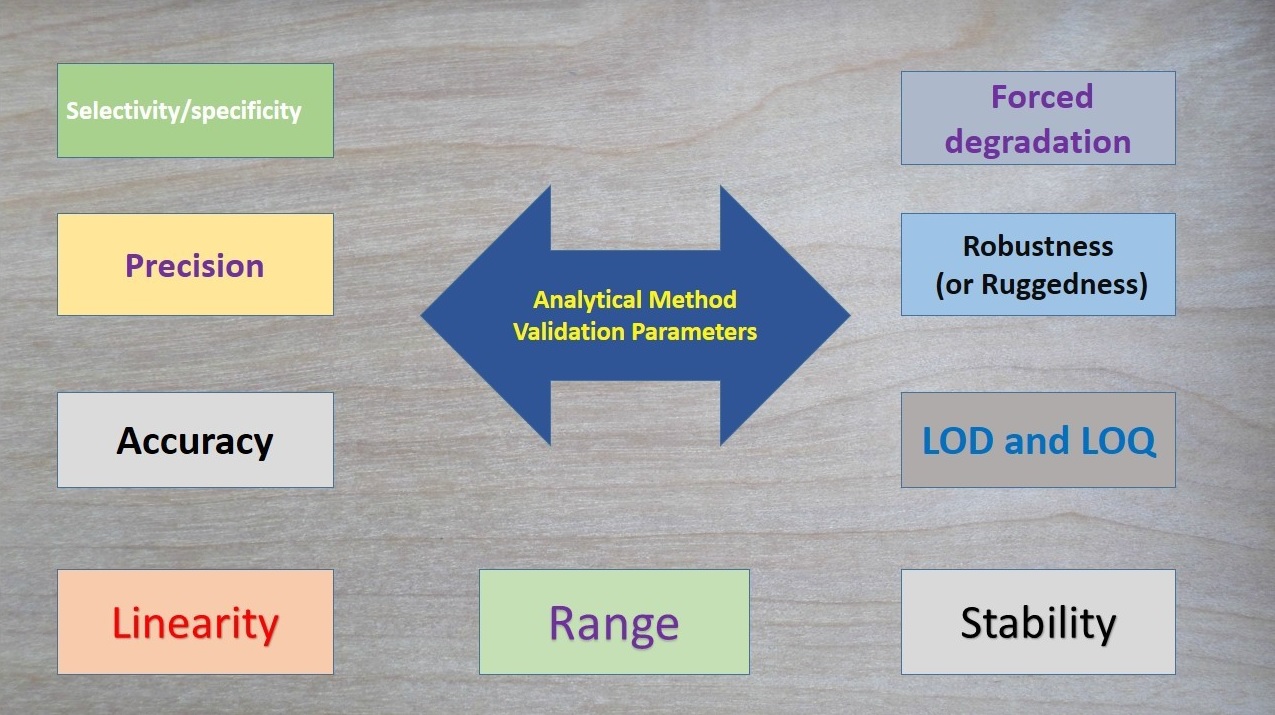
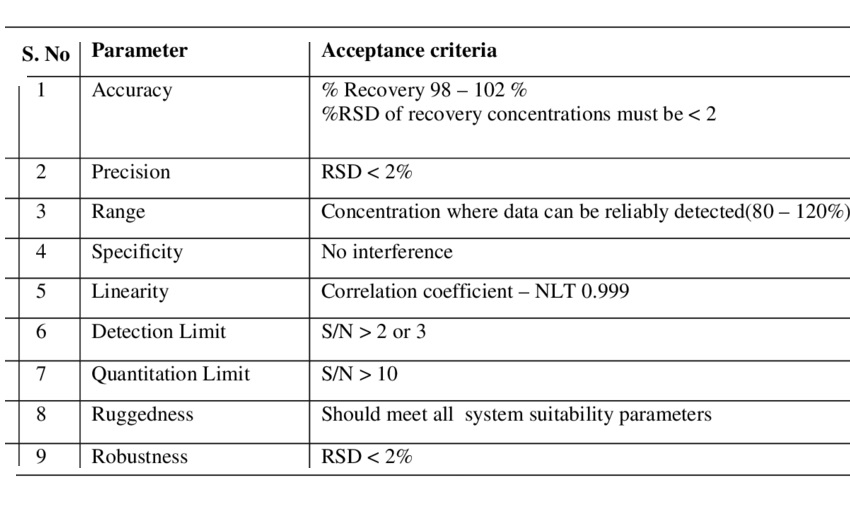
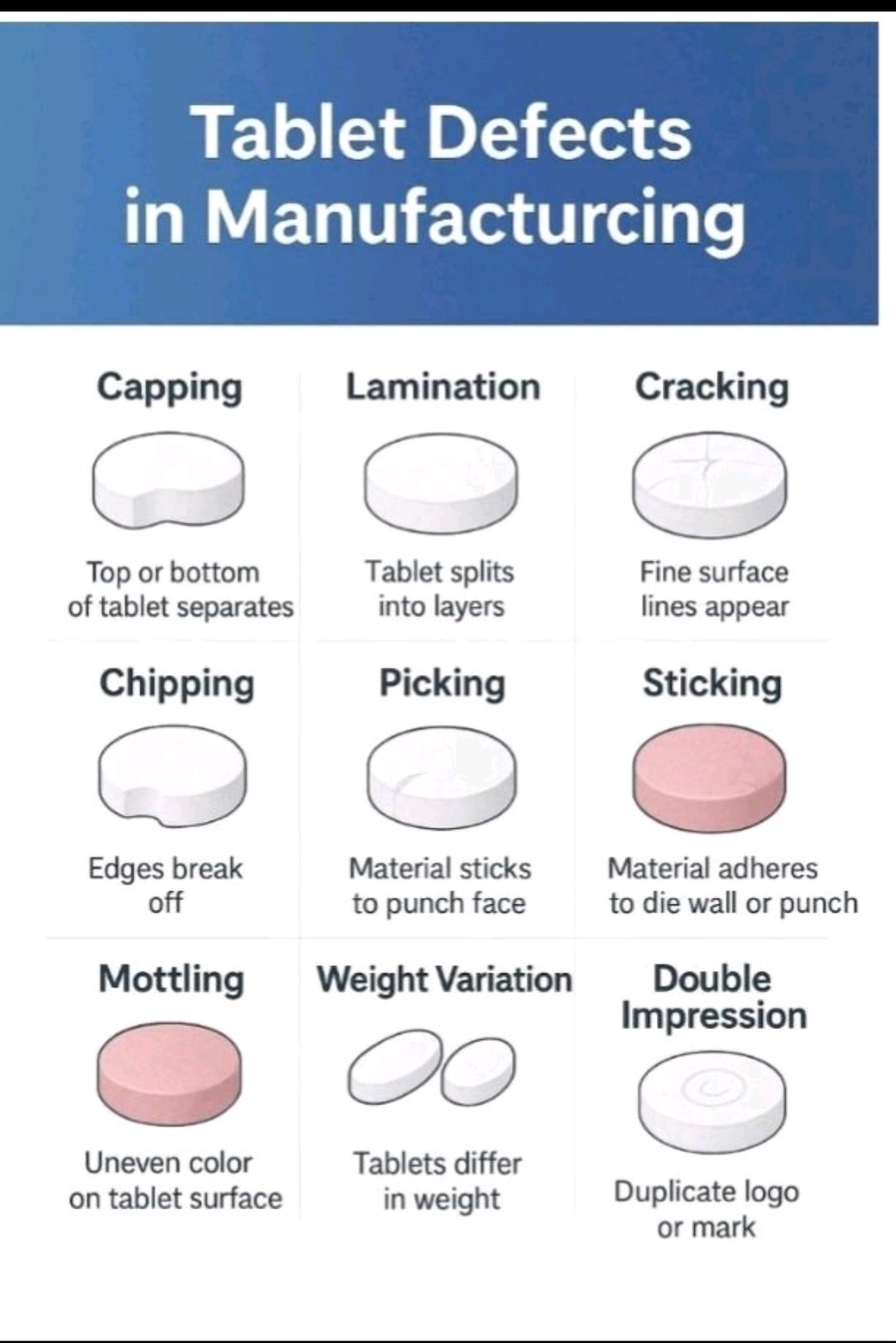
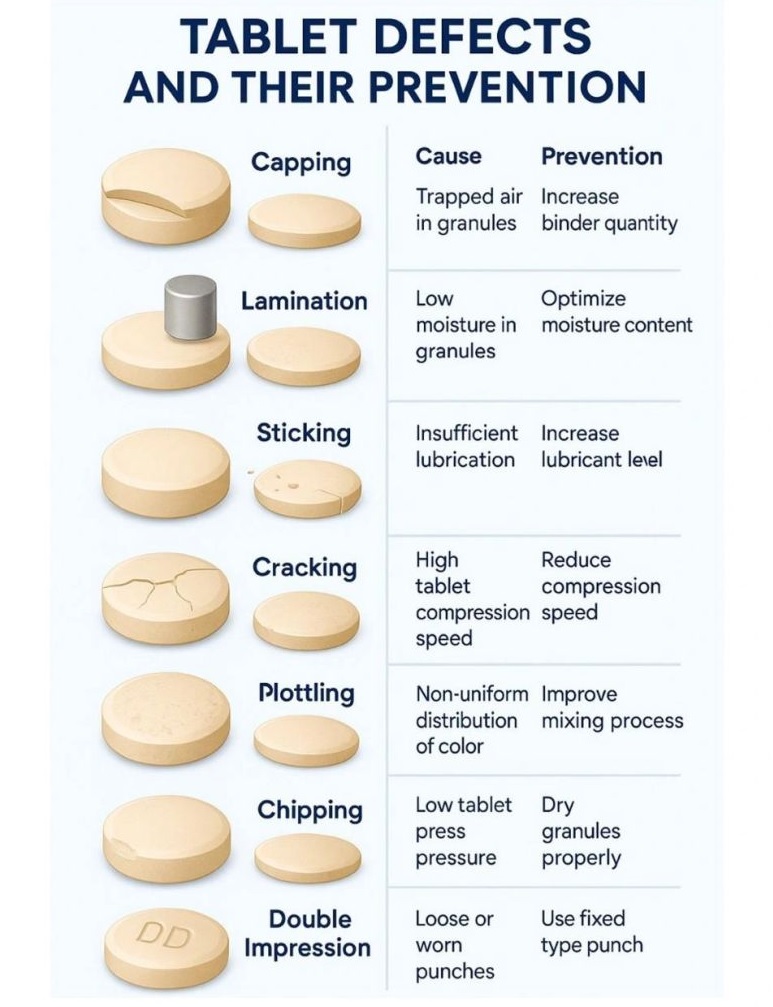
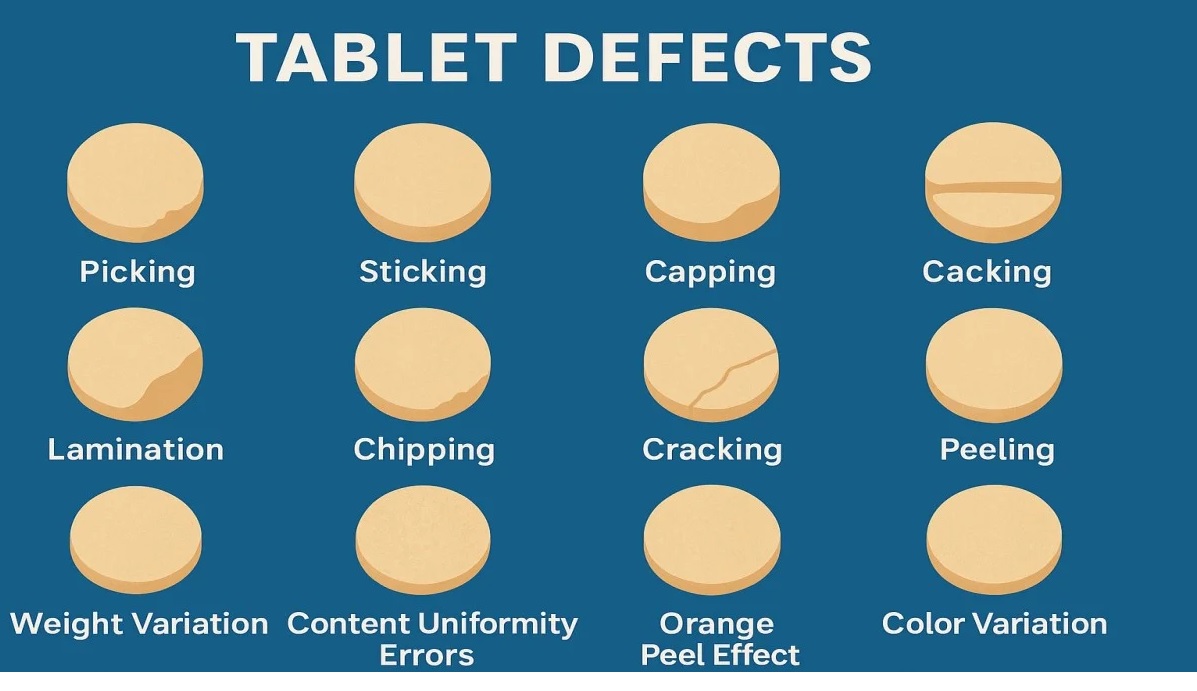
Tablet defects are common issues in pharma manufacturing, including compression issues (capping, lamination, cracking, softness), tooling/material adhesion (sticking, picking, binding), appearance problems (mottling, color variation, rough surface, double impressions, shape issues, coating flaws like blistering/pitting), and weight/size variations (uneven thickness, incorrect dimensions).
Relative Response Factor (RRF) formula compares the detector response (usually peak area) of an impurity to the main Active Pharmaceutical Ingredient (API) at equal concentrations, crucial for accurate impurity quantification; the core formula is
RRF = Response(Impurity) / Response(API),
often expressed as;
RRF = (Area Impurity / Area API) × (Conc API / Conc Impurity)

Pharmacovigilance (PV) is the critical science and activity of monitoring medicine and vaccine safety after they are on the market, focusing on detecting, assessing, understanding, and preventing adverse drug reactions (ADRs) or any other drug-related problems to ensure patient safety and maximize benefits. It involves a continuous process of gathering reports, analyzing data, and taking action to manage risks, involving patients, healthcare providers, manufacturers, and regulators in a shared responsibility for public health.

Analytical Method Validation
Solution stability tests how a drug behaves under normal storage (room temp, fridge, light protection) over time, while
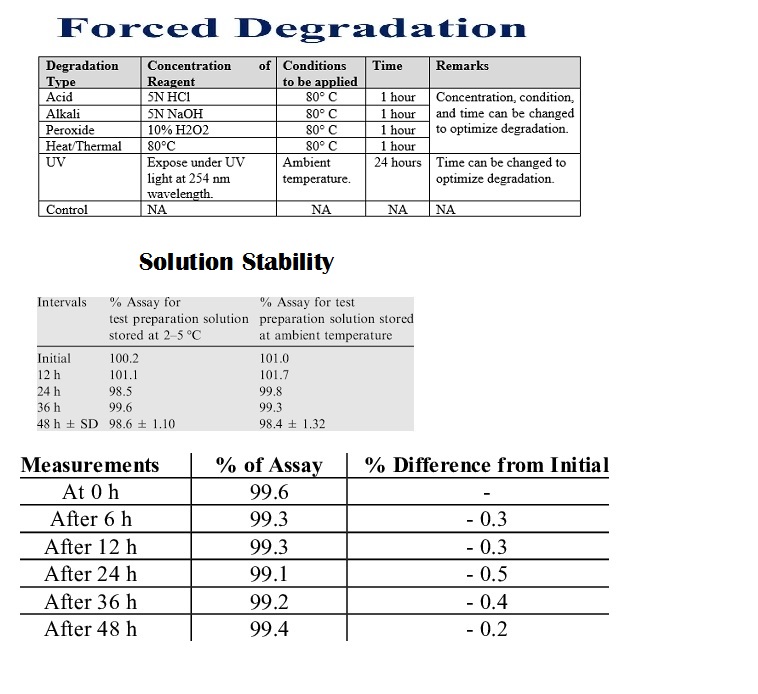
Forced degradation (stress testing) uses extreme conditions (acid/base, heat, oxidation, light) to rapidly identify potential breakdown products, establish degradation pathways, and ensure the analytical method can separate the main drug from these impurities, proving it’s “stability-indicating”
Solution stability acceptance criteria in analytical validation ensure prepared standards and samples remain consistent over time, typically requiring the assay result (e.g., % purity) to stay within ±2% of the initial value, with impurity levels controlled within tighter ranges (e.g., ±0.04% total) for a defined period (e.g., hours to days at room or refrigerated temp).
prepare standard/sample solutions, store aliquots under relevant conditions (room temp, fridge, light protection), and analyze them at time zero and various intervals (e.g., 2,4,6,12, 24, 48 hrs) to check for degradation, ensuring results stay within predefined limits (e.g., ±2-5%) to establish the solution’s shelf-life for routine use.


Analytical Method Validation
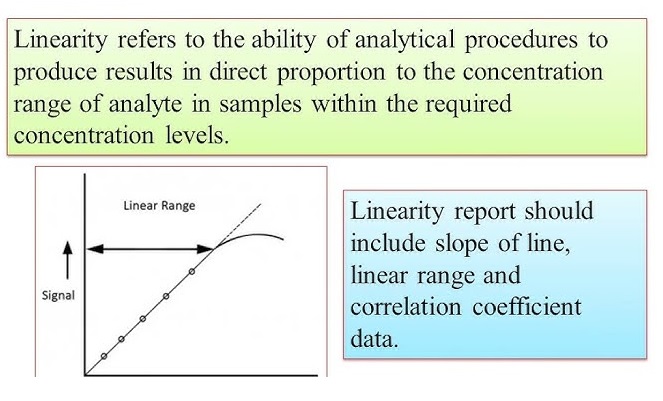
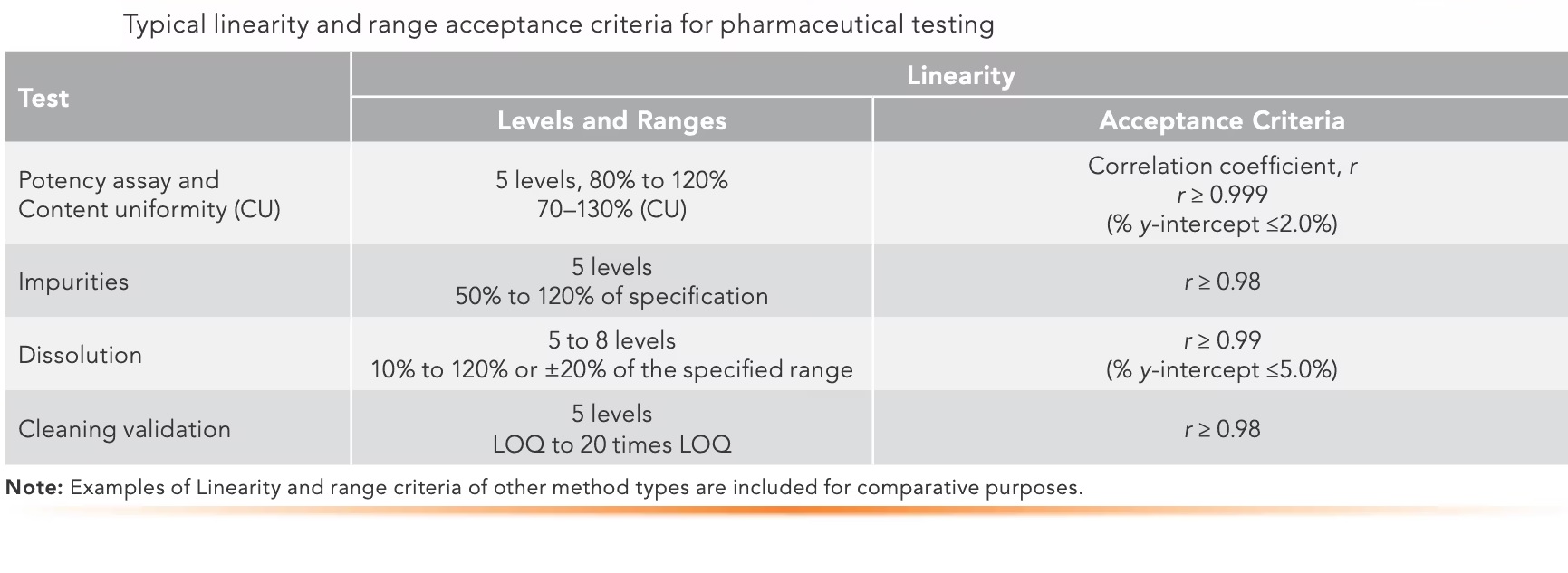
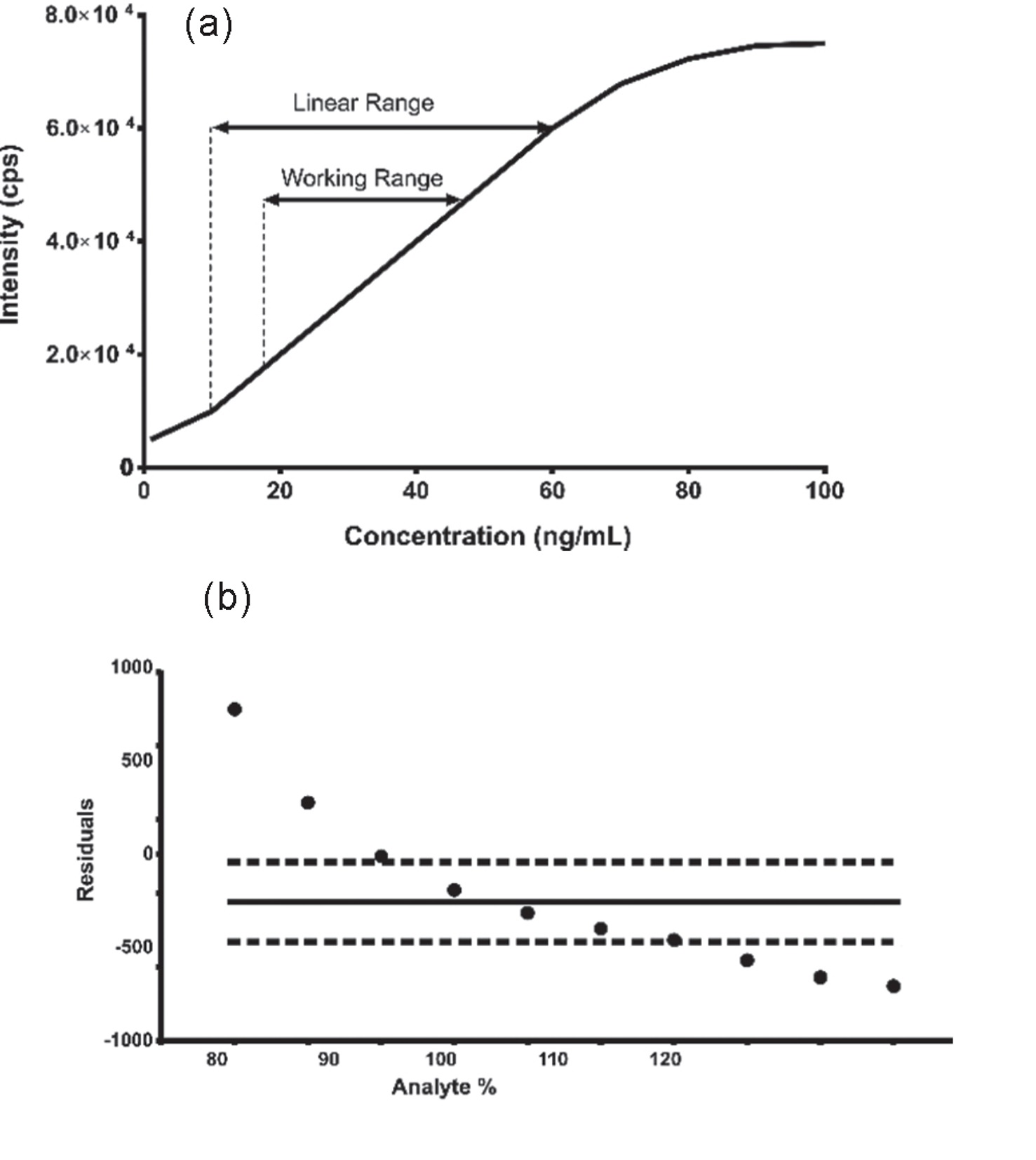
Linearity proves results are directly proportional to analyte concentration (a straight line on a graph),while
Range defines the concentration interval where the method reliably shows good precision, accuracy, and linearity, typically from low to high levels, ensuring fitness for purpose. Linearity is checked with calibration curves (𝑟2>0.995) and visual inspection, while the range confirms the method’s ability to measure accurately across expected sample variations.
In Linearity test a set of standard solutions containing the API at various concentrations e.g., 10%, 25%, 50%, 75%, 100%, 120% and 150 % of the target concentration is prepared.
The range of an analytical method typically includes the lowest and highest levels of the target analyte that the method is expected to measure accurately.
Analytical Method Validation
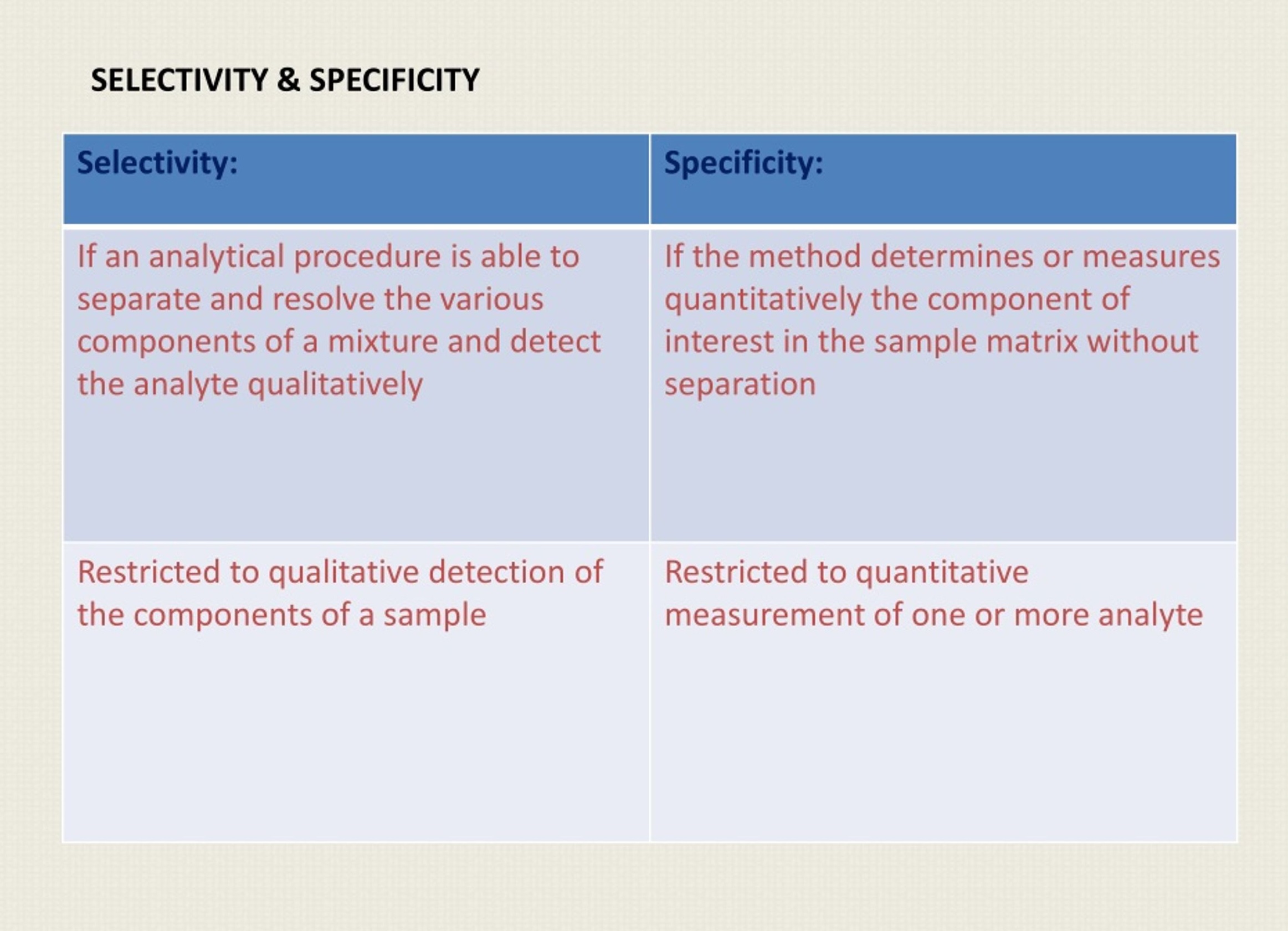
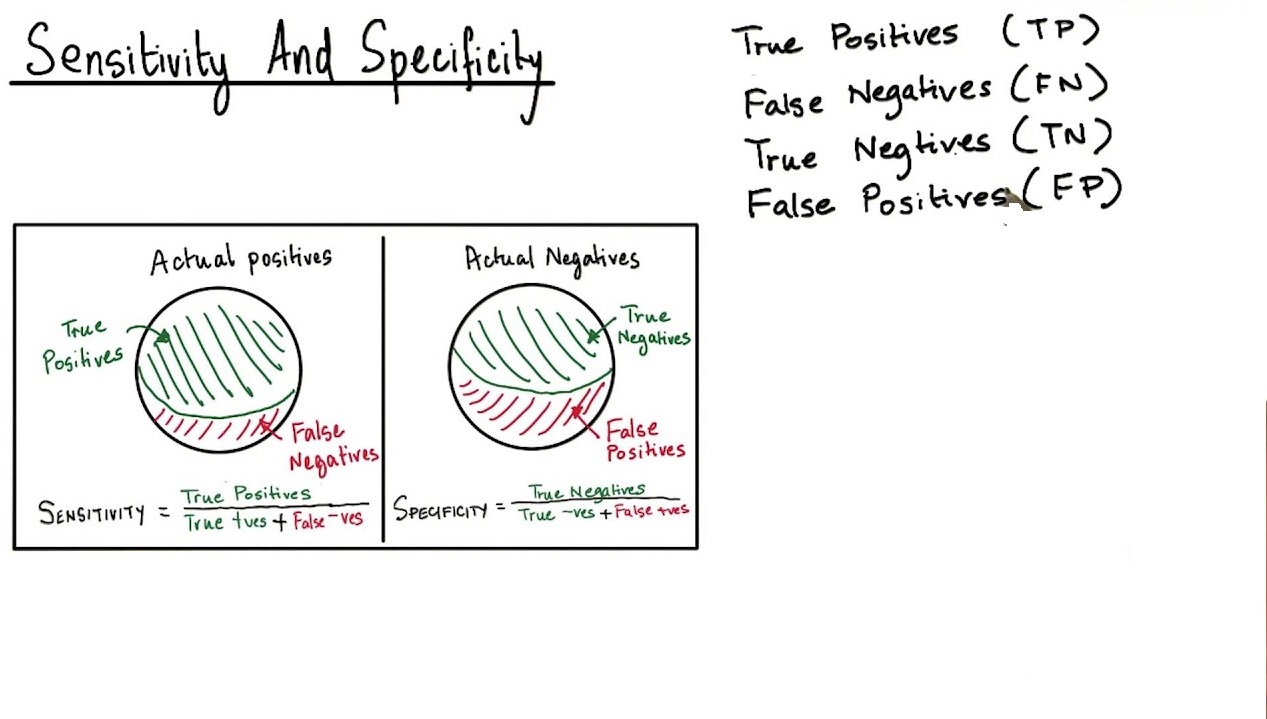
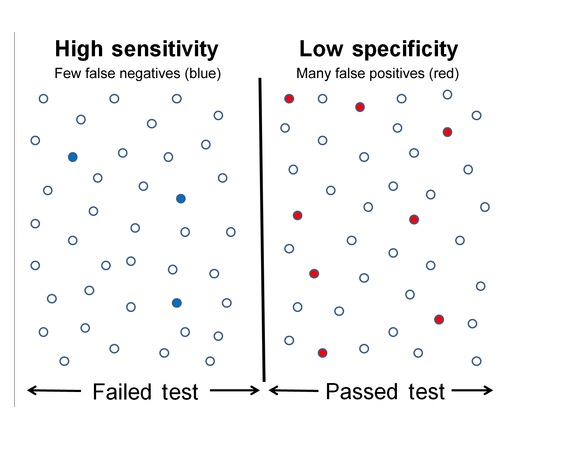
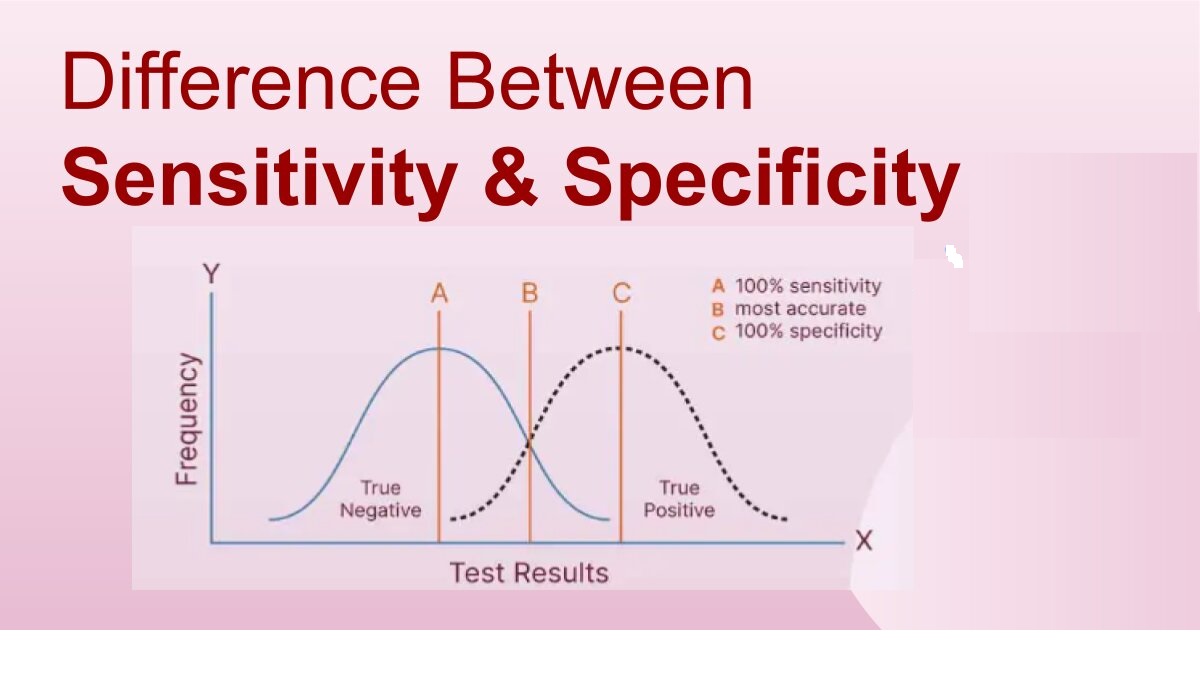
Specificity is the ability to measure the target analyte without interference from other components (impurities, degradants, matrix), while sensitivity refers to the method’s ability to detect and accurately quantify small amounts of the analyte, often defined by the Limit of Detection (LOD) and Limit of Quantitation (LOQ); and Selectivity (often related to Specificity but broader in analytical chemistry) is a method’s ability to measure one target analyte without interference from others.

 (𝑟2>0.995) and visual inspection, while the range confirms the method’s ability to measure accurately across expected sample variations.
(𝑟2>0.995) and visual inspection, while the range confirms the method’s ability to measure accurately across expected sample variations.