
Title:-Interview Questions
- What are difference between calibration & validation & qualification?
Calibration – The set of operations that establish under specified conditions the relationship between values indicated by an instrument or system for measuring and corresponding values of reference standard.
Validation- Action of proving & documenting that any process actually & consistently leads to expected results.
Qualification – Action of proving & documenting that any premises, systems & equipments are correctly installed & lead to expected results.
2. Define CAPA?
Corrective and preventive actions
3. What is the difference between drug purity & potency?
Purity: it is the absence of unwanted substances like impurities & contaminants
Potency: it is a measure of drug activity measured in terms of amount of drug required to produce an effect.
4. Which gases are used in GC?
Helium & Nitrogen.
5. What is the difference b/w temp change control & deviation?
Change control: it’s planned/permanent change after assessing the impact on other functions.
Deviation: unplanned/Temporary change.
6. What is friability?
Ans: The percentage of loss mass of a solid dosage form after 100 times revelation in 4 minutes due to friabilizer is called Friability.
7. What is Hardness?
Ans:Required energy for break a Solid dosages form on their edge.
8. What is pH?
Ans: pH/pOH is the negative logarithm of Hydrogen ion/Hydroxyl ion concentration. Measurement of pH of any aqueous sample solution gives the degree of acidity or alkalinity of the solution. Mathematically it may be represented as follows;
pH= – log [ H+ ].
pOH = – log [OH- ]
9. Why pH is important in pharmaceuticals?
Ans: pH is important in pharmaceuticals because some active ingredient is stable in one pH and degrades in another
pH. So in-case of product stability pH is an important factor.
10. What is Dissolution?
Ans: The process by which the solid particle or medicament are dissolve in suitable solvent for ease of absorption.
11. What is difference between Dissolution and Assay?
Ans: Dissolution- is the amount of drug released in a certain period of time from a dosage form.
Assay- is the amount of drug that is contained by a unit dosage form of medicament.
12. What is disintegration? Importance in pharmaceuticals dosage form?
Ans: Breakdown of unit solid dosage form or larger granules into smaller one (smaller than 2 mm).
In most cases dissolution is directly proportional to the disintegration. More faster the solid dosage form disintegrates, more faster it
dissolve and goes into the systemic circulation and gives the therapeutic response.
13. What do you mean by DQ, IQ, OQ, & PQ ?
Design Qualification (DQ): documented verification that the proposed design of the facilities, equipment, or systems is
suitable for the intended purpose.
Installation Qualification (IQ): documented verification that the equipment or systems are installed or modified &
comply with the approved design of the manufacturer’s recommendations and/or user requirements.
Operational Qualification (OQ): documented verification that the equipment or systems are installed or modified &
perform as intended throughout the anticipated operating ranges.
Performance Qualification (PQ): documented verification that the equipment and ancillary systems are connected &
can perform effectively and reproducibly based on the approved process method and specifications.
14. Define complaint & recall?
All quality related complaints of a product from the market should be recorded & investigated accordingly to written procedure.
Recall is the removal from the market of specified batches or all batches of a product.
15.Define Corrective Action & Preventive Action (CAPA) ?
Corrective Action : Action taken to rectify, fix or correct a specific deviation, defect or undesirable situation.
Preventive Action : Action taken to eliminate the cause of deviation, defect, or other undesirable situation in order to prevent the future occurrence of such or similar an event.
16. What is self inspection?
Self inspections is conducted by internal team in order to monitor the implementation and compliance with Good Manufacturing Practice principles and to propose necessary corrective measures.
17. Define Stating Materials, Intermediate , Bulk & Finished Products ?
Stating Materials : A starting material is known as raw material or an API. A substance is a defined quality which used in production of a pharmaceutical product & non-pharmaceutical product excluding packaging material.
Intermediate : A partly processed material that must undergo the further manufacturing steps before a bulk product.
Bulk Product : Any product that has completed all processing steps up to but not including final packaging.
Finished Products : A medicinal product, which has undergone all stages of manufacture including packaging.
18. What is the differences between documents & records?
Documents : Document is a controlled written procedure, policy, forms or other pieces of paper as defines a company requirement. & describe how and what to do.
Records : When data or results providing evidence of activities performed then it is recorded.
19.What is HVAC system & its functions?
Heating, Ventilating and Air Conditioning system is used for temperature and humidity control with in a manufacturing
environment.
20. What is SOP? Why it is needed ?
SOP (Standard Operating Procedure): A written approved procedure which gives instruction for performing operation/task not necessarily specific to a given product or material but of a more general matter.
21. What is Clean Room?
A room in which the concentration of airborne particles is controlled to meet specified airborne particulate cleanliness class.
22. What is Aseptic Area?
Any area is an aseptic for which airborne particulate and microorganism levels are controlled to specific levels appropriate to the activities conducted within that environment.
23. What is Air lock?
Airlock: An enclosed space with two or more doors, which is interposed between two or more rooms.
24. What Is Site Master File ?
Site Master File is prepared by the manufacturer and contains specific information about the quality assurance, the production and/ or quality control of pharmaceuticals manufacturing operations carried out at the named site and any closely integrated operations at adjacent and nearby buildings. It only part of a pharmaceuticals operation is carried out
on the site.
25. What are the differences between Sanitization and Sterilization?
Sanitization:
Reduction the number of micro-organism to a safe or relatively safe level as determined by applicable, regulations or
the purpose of application
Sterilization: Sterilization is a process or technique to remove bacteria and all types of viable microorganism.
26. What are the parameters performed during Analytical Method validation?
Accuracy, Precision, Specificity, Detection Limit, Quantitation Limit, Linearity & Range, Ruggedness & System suitability Testing.
27. what is HEPA filters?
High Efficiency Particulate Air .High Efficiency Particular Air Filter (HEPA) is a air filtering system. HEPA filter capable of retaining 99.97 percent of particles as small as 0.3 μm.
28. what is DOP?
DOP : Dioctyl Phthalate .Dioctyl Phthalate test is used to determine the integrity of HEPA filters.
29. What is difference between QA and QC ?
Quality Assurance(QA) is the section made with the objective of ensuring that pharmaceutical products are of the quality required for their intended use .QA therefore incorporates GMP and other factors such as product design and development, GLP and GCP.
Quality Control (QC) is the section where the product is actually tested for expected behavior or standards as per defined specifications.
30. What is Contamination?
An undesired introduction of impurities of a chemical or microbiological nature ,or of foreign matter, into or on to a starting material or intermediate during production , sampling , packaging or repackaging, storage or transport.
31. What is the composition of a C18 column?
Octadecyl carbon chain bonded with silica.
32. What do you mean by LOD and water content?
LOD is the loss during drying of sample as per prescribed conditions which gives loss (presence) of all evaporating solvents along with water.It is the dry base.
Water Content gives the moisture present in the sample only. It is anhydrous base.
33. Why we use toluene for resolution in UV calibration?
Toluene gives Maxima and Minima at 266 nm and 269nm to check resolution in UV calibration. Hexane used as a diluents because it has no UV absorbance.
34. Why we use disodium tartare for determination of factor in Karl Fischer titration?
Disodium Tartrate is used as a primary standard substance for the standardization because of the following properties:
1. Available in a stable crystalline hydrate form.
2. Its water content is close to theoretical value.
3. Substances in nearly 100% purity.
4. More favorable gravimetric factors.
5. Stable at extreme humidity.
6. It is relatively insoluble in absolute methanol
7. Its water content is rapidly and quantitatively titrated to a sharp end point.
35. What are closely monitor parameters in stability study? Temperature and Relative Humidity.
36. What does u mean by MACO & NOEL level?
MACO-Maximum Allowable Carryover
NOEL-No Observed Effect level
both terms are used in cleaning validation to set limits.
37. What is Process validation?
Establishing a documented evidence with high degree of precision that specific process will consistently produce a product which meets its predetermined specifications & quality parameters.
38. What are stability zones & climatic conditions?
Zone-1:(Temperate Region)
Great Britain/north Europe/Canada/Russia (Temp-21/ RH-45%)
Zone-2: (Subtropical/Mediterranean Region)
USA/Japan/South Europe (Temp-25/ RH-60%)
Zone-3: (Hot & Dry Region)
Iran/Iraq/Sudan/India (Temp- 30/ RH- 55%)
Zone-4: (Hot & Humid Region )
Brazil/Ghana/Nicaragua/Philippines (Temp-30/ RH -70%)
39. What is photo stability?
It was study performed to evaluate & demonstrate that light exposure doesn’t result in unacceptable change in new drug substances.
40. What is the wave length of polarimeter lamp?
589.3 nm.
41. Which gases are used in Gas Chromatography?
Helium, Hydrogen & Nitrogen.
42. What is dead leg?
A dead leg is defined as an area in a piping system where liquid can become stagnant and not be exchanged during flushing.
43. How many Tablets shall be taken for checking friability?
For tablets with unit mass equal or less than 650 mg shall take sample of whole tablets corresponding to 6.5g.
For tablets with unit mass more than 650 mg shall take a sample of 10 whole tablets.
44. What is the formula for calculating weight loss during friability test?
%Weight Loss = Initial Weight – Final Weight X 100_____ Initial Weight
45. Why do we calibrate a qualified equipment/instrument on definite intervals?
An equipment or instrument can ‘drift’ out of accuracy between the time of qualification and actual use. So it is recommended to calibrate and recalibrate the measuring devices and instruments on predetermined time intervals, to gain confidence on the accuracy of the data.
46. Why do we consider three consecutive runs/batches for process validation?
The number of batches produced in the validation exercise should be sufficient to allow the normal extent of variation and trends to be established and to provide sufficient data for evaluation and reproducibility. · First batch quality is accidental , · Second batch quality is reproducible , · Third batch quality is validation (confirmation).
47. What is bracketing & matrixing in stability testing?
Both Matrixing & Bracketing’s are reduced stability testing designs Bracketing The design of a stability schedule, such that only samples of extremes of certain design factors (ex:strength,package size) are tested at all time points as in full design.The designs assumes that the stability of any intermediate level is represented by the stability of extremes tested. Matrixing The design of a stability schedule, such that a selected subset of possible samples for all factor combinations is tested at a specified time point.At a subsequent time point another subset of samples for all factor combination is tested. The design assumes that the stability of each subset samples tested represents the stability of all samples at a given time point. There for a given time point other than initial & final ones not every batch on stability needs to be tested.
48. What is Capping in Tablets?
It is partial or complete separation of the top or bottom of tablet due air-entrapment in the granular material.
49. What is Lamination in tablets?
It is separation of tablet into two or more layers due to air-entrapment in the granular material.
Is the separation of a tablet into two or more distinct horizontal layers.
50. What is Chipping in tablets?
• Chipping‘is defined as the breaking of tablet edges, while the tablet leaves the press or during subsequent handling and coating operations.
51. What is CRACKING in Tablets?
Small, fine cracks observed on the upper and lower central surface of tablets, or very rarely on the sidewall are referred as Cracks.
52. What is MOTTLING in Tablets?
Mottling‘is the term used to describe an unequal distribution of colour on a tablet.
53. What are the recommended storage conditions for empty hard gelatin capsules?
Ans: Temperature;15 – 25 C & Humidity;35 -55% RH.
54. What is Limit of detection (LOD)?
The lowest amount of analyte in a sample which can be detected but not quantitated as an exact value.
55. What is Limit of quantitation (LOQ)?
The quantitation limit of an individual analytical procedure is the lowest amount of analyte in a sample which can be quantitatively determined with suitable precision and accuracy.
56. What is Precision?
The precision of an analytical procedure expresses the closeness of agreement (degree of scatter) between a series of measurements obtained from multiple sampling of the same homogeneous sample under the prescribed conditions.
57. What is Repeatability?
Repeatability expresses the precision under same conditions: same analyst, same apparatus, short interval of time, identical reagents.
58. What is Reproducibility?
The reproducibility expresses the precision under different conditions for instance: laboratories, reagents from different sources, analysts, days, apparatus from different manufacturers, etc.
59. What is Ruggedness?
The degree of reproducibility of test results obtained by the analysis of the same samples under change in equipment, laboratory, and analyst.
60. What is Robustness?
Robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate, variations in method parameters and provides an indication of its reliability during normal use such as different temperatures, mobile phase compositions, flow rates, or injection volumes etc.
61. What is Specificity?
Specificity is the ability to assess unequivocally the analyte in the presence of components which may be expected to be present. Typically these might include impurities, degradants, placebo matrix, etc.
62. Expiry date – The date given on the individual container (usually on the label) of a drug product up to and including which the product is expected to remain within specifications, if stored correctly.
63. Retest date – The date when a material should be re-examined to ensure that it is still suitable for use.
64. What is Bioburden?
The total number of microorganisms associated with a specific item prior to sterilization.
65. Colony Forming Unit (CFU):-
A microbiological term that describes the formation of a single macroscopic colony after the introduction of one or more microorganisms to microbiological growth media. One colony forming unit is expressed as 1 CFU.
66. Decontamination:-
A process that eliminates viable Bioburden via use of sporicidal chemical agents.
67. Disinfection:-
Process by which surface Bioburden is reduced to a safe level or eliminated.
68. Component: Component means any ingredient intended for use in the manufacture of a drug product including those that may not appear in such drug product.
69. Drug – Product: ‘Drug Product’ means a finished dosage forms (e.g. Tablet, Capsule, Solution etc.) that contains an active drug ingredient generally but not necessarily in association with inactive ingredients.
70. Active Ingredients: Active ingredient means any component that is intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment or prevention of disease or to effect the structure of any function of the body of man or other animals.
71. Excipient: Excipients are the additives/no drug component used to convert pharmacologically active compounds in a pharmaceutical dosage forms suitable for administration of patients.
72. Inactive Ingredient: ‘Inactive ingredient’ means any component other than an active ingredient.
73. Bulk Product: Any products, which has completed all processing stages up to, but not including final packing. Any processed product, which still has to undergo the packing operation in order to become a
finished product.
74. Finished Product: A medicinal product, which has undergo all stages of manufacturing operations including packaging in its final container and labeling.
75. Strength: Strength means the concentration of the drug substances (for e.g. weight/ weight, weight / volume or unit dose and or the potency of a drug product).
76. Potency: Potency means the therapeutic activity of the drug product as indicated by appropriate laboratory tests or by adequately developed and controlled clinical data.
77. Efficacy: It is a term used to know the response of any drug produced whether optimum or not.
78. What is Theoretical Yield?
The quantity that supposed to be obtained at any appropriate phase of manufacturing, processing or packing of a particular drug product, based upon the quantity of components to be used, in absence of any loss or error in actual production.
79. What is Actual Yield?
‘Actual Yield’ means the quantity that is actually produced at any appropriate phase of manufacture, processing or packing of a particular drug product.
80. What is OTC Drug?
OTC Drug: Over the Counter Drug. OTC drugs are those drugs which can be used without the prescription of a registered physician.
81. Optical Rotation: The optical rotation of a substance is the angle through which the plane of polarization is rotated, when polarized light is passed through a chemical substance.
Dextro-rotation is designated (+) and Levo-rotation is designated (-).
82. Fumigation: Fumigation is the process to reduce microbial load in a particular area by using poisonous fumes or gases to destroy living organisms.
83. Accuracy: The closeness of the result obtained during measurement or analysis to the its true value.
84. Conductivity:
Conductivity is the measurement of materials ability to conduct electric current. Measurement of conductivity of sample solution gives the degree of electrical conductance in a defined volume of the solution.
85. Potable Water: Potable water is drinking water, treated by the local authority or an site, which is suitable for human consumption.
86. DM Water: DM Water stands for Demineralized water. The water free from minerals is called Demineralized water.
87. Water For Injection: Water for injection is intended to a high degree of chemical purity and to be free from pyrogenic
substances and endotoxin.
88. TDS: TDS means Total dissolve solid. The amount of solid present in dissolved state in DM or Tape water.
89. Pyrogen: In Greek word “Pyrogen” means “Production of fever” any substance that produces a fever.
90. LAL: LAL stands for “Limulus Amoebocyte Lysate” test. This test determines the presence or the concentration of endotoxin in products.
91. Endotoxin: Bacterial Endotoxin or pyrogen is metabolic products of living micro-organism or the dead micro-organism causing a specific pyretic response upon injection.
92. Positive Controls: Positive controls is a culture medium which is used for sterility test is checked for its efficacy to support growth of different organism by adding 10-100 cfu of m gm. This is known as positive control.
93. Negative Controls: Negative controls are the culture media without addition of microbial culture. The –ve control is used to verify the sterility of the medium before, during and after the incubation period of the sterility tests.
94. ATCC (American Type Culture Collection): It is a institution which gives a specific number to different Micro organisms based on the variation of the strain.
95. Positive Pressure: Positive pressure are the pressure created in the aseptic production area to avoid contaminated air pass in.
96. Differential Pressure: Differential Pressure means the pressure difference between two adjacent rooms.
97. Suspension: A suspension is a preparation of finely divided undissolved drugs, dispensed in a liquid phase. It is a two phase dosage system in which a finely divided solid in mixed in desire liquid.
98. Emulsion: Emulsion are usually liquid preparation which are prepared by addition emulsifying agent and purified water to liquid drugs.
99. Suppository: Suppositories are solid preparation intended to insertion in to the rectum, urethra or vaginal cavity. Suppositories are usually prepared by molding uniform mixers of drugs and bases in to a suitable shape. Suppositories melt or soften at body temperature or dissolve slowly in the secretions. Suppositories are usually prepared by by mixing drugs with fat-type bases water-miscible bases or other suitable materials.
100. Ointment: Ointment are usually homogeneous, semisolid preparations for external application that they may be applied to the skin .
101. Cream: Semisolid emulsion system with opaque appearance. It may be a water in-oil or oil in – water type.
102. RSD (Relative Standard Deviation):
Relative Standard Deviation is given by:
%RSD = (S/X)*100
S= Standard Deviation, X=Mean value.
103. What is Form 482?
FDA may conduct an inspection of your operation for a variety of reasons, such as a routinely scheduled investigation, a survey, or a response to a reported problem. The investigator will present credentials and “Notice of Inspection” upon arriving at your plant.
104. What is Form 483?
An FDA form 483 is issued to firm management at the conclusion of an inspection when an investigator has observed any conditions that in their judgement may constitute violations of the Food Drug and Cosmetic (FD&C) Act and related acts.
105. Out of Specifications (OOS): Results generated during testing that do not comply with relevant standard or specification.
106. Out of Trends (OOT): Any test result obtained for a particular batch that is markedly different from the results of batches in a series.
107. What is Root Cause Analysis?
Root cause analysis is a problem solving technique for identifying the basic or root cause that underlie the occurrence or possible occurrences of an adverse event in a process similar to diagnosis of disease – with the goal always in mind of preventing re-occurrence.
108. Risk Assessment: A systematic process of organizing information to support a risk decision to be made within a risk management process. It consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards.
109. Prospective validation: The execution and documentation of pre-approved test protocol, which is designed to prove that a process performs as intended, prior to the release of a manufactured product for distribution. This Validation carried out during the development stage by means of a risk analysis of the production process.
110. Concurrent Validation: Validation carried out during routine production of products intended for sale.
111. Retrospective Validation: It involves the examination of past experience of production on the assumption that composition, procedures, and equipment remain unchanged The evaluation determines the reliability, accuracy, and completeness of a system.
112. what is Audit Trail?
The audit trail is the form of metadata containing information associated with every action that relate to the creation, modification or deletion of GxP records.
113. Critical Process Parameters (CPP): A Process parameter whose variability has an impact on a critical Quality Attributes and thereof should be monitored or controlled to ensure the process produces the desired quality.
114. Critical Quality Attributes (CQA): A Critical Quality Attribute is a physical, chemical, biological or microbiological property or characteristics that should be within an appropriate limit, range or distribution to ensure the desired product quality.
115.Calibration: It is a demonstration that, a particular Instrument or device produces results with in specified limits by comparisons with those produced by a reference or traceable standard over an appropriate range of measurements.
116. What are the Good Manufacturing Practice (GMP) Guidelines?
ICH: Q7 Good Manufacturing Practice
US FDA: 21 CFR Part 210 – cGMP General
211- cGMP for Finished Pharmaceuticals
WHO- Technical Report Series-937
PIC/S- Recommendations PE-009
EU-Part1, Annex5
EU-Part11, Chapter 13
117. What is Molarity ?
- Definition: Number of Moles of substance dissolve in per liter of solution.
- Calculation:
Molarity= Number of Moles of substance__
Volume of solution in liter
118. What is Normality ?
- Definition: Normality is the number of equivalents of a reactive species per liter of solution.
- Calculation:
Normality= Molarity x n
n=is the number of equivalents per mole.
119. What is Sampling?
Sampling is a process of collecting a small portion of material from a bulk of material received in the form of consignment or whole batch.
120. Primary Packing Materials: Materials which comes in direct contact with the drug product. Eg. PVC, Foils.
121. Secondary Packing Materials: Materials which comes in direct contact with primary packing materials. Eg. Cartons, leaflets.
122. Tertiary Packing Materials: Materials which comes in direct contact with secondary packing materials. Eg. Corrugated boxes.
123. Minor defects: A defect that does not affect product safety, purity or identity or package integrity of function.
124. Major defects: A defect that jeopardizes the integrity or function of the package
125. Critical defects: A defect that can compromise product safety, purity or identity that may be harmful to the consumer.
126. What is Hazardous waste?
Waste that is unwanted, unusable and dangerous or potentially harmful to our health or the environment.
127. What is “significant change” in Stability Study?
“significant change” occurs at any time during 6 months testing at the accelerated storage condition.
In general, “significant change” for a drug product is defined as:
- A 5% change in assay from its initial value; or failure to meet the acceptance criteria for potency when using biological or immunological procedures.
2. Any degradation product’s exceeding its acceptance criterion.
3. Failure to meet the acceptance criteria for appearance, physical attributes, and functionality test (e.g., color, phase separation, resuspendability, caking, hardness, dose delivery per actuation); however, some changes in physical attributes (e.g. melting of creams) may be expected under accelerated conditions; and, as appropriate for the dosage form.
4. Failure to meet the acceptance criterion for pH.
5. Failure to meet the acceptance criteria for dissolution testing at 12 dosage unit.
128. Reagent: Reagent is a substance or compound that is added to a system in order to bring about a chemical reaction, or added to see if a reaction occurs.
129. Indicators: Indicators are reagents used to determine the specified endpoint in a chemical reaction which indicates the change in colour, to measure hydrogen-ion concentration (pH) or to indicate the desired change in pH has been effected.
130. Buffer solutions: Buffer solutions are solutions that resist change in the activity of an ion on the addition of substances that are expected to change the activity of that ion.
131. What is Carbon Dioxide-Free water
It is the purified water that has been boiled vigorously for 5 minutes or more and allowed to cool while protected from absorption of carbon dioxide from the atmosphere.
132. Molar solutions (M): Molar solutions are solutions that contain 1 gram molecule of the compound in 1 litre of solution.
133. Normal solutions (N): Normal solutions are solutions that contain 1 gram equivalent weight of the compound in 1 litre of solution.
134. Percentage weight in weight (%w/w): It expresses the number of grams of solute in 100 g of product.
135. Percentage weight in volume (% w/v): It expresses the number of grams of solute in 100 ml of product.
136. Percentage volume in volume (% v/v): It expresses the number of milliliters of solute in 100 ml of product.
137. Percentage volume in weight (% v/w): It expresses the number of millilitres of solute in 100 g of product.
138. Parts per million (ppm): “Parts per Million” is a way to quantify very low concentrations of substances. For example, 1 ppm is equivalent to 1 milligram of solute per litre of liquid (abbreviated as mg/L). 1 milligram of solute per millilitre of liquid (abbreviated as mg/ml).
1 mg/ml = 1000 ppm
139. What is Cleaning Validation?
A methodology used to assure effectiveness and consistency of cleaning process to remove residue and contaminants from equipment.
It guarantees that manufacturing equipment is clean and free from contaminants before using for new product batch.
140. Type A Cleaning: It is dry cleaning process performed between the same type of products or campaign batches generally called as batch to batch changeover. This type of cleaning consists of removal of drug powders from the surface with the help of clean cloth or scraper/scrubber.
141. Type B Cleaning: It is wet cleaning process in which cleaning solution is used for cleaning of Equipment during product to product changeover.This type of cleaning consists of total removal of drug residue from the surface with the help of cleaning agent and purified water to avoid previous product residue (contamination) in next product.
142. Rinse Sampling: It is the method by which all the contact surface of the cleaned equipment is rinsed with specified amount of water then collecting certain volume of the rinse water into a vial for analysis.
143. Swab Sampling:This method involves wiping a predetermined area of the cleaned equipment with a swab that has been moistened with a solvent determined by the contaminating compound. Usually the surface is wiped with one side of the cotton swab using a certain number of strokes, then the swab is flipped and wiped with other side of the cotton swab.
144. Flammable Chemicals: A flammable material can be a solid, liquid or gas. It is defined as any liquid having a flash point below 100o F (37.8oC). Example: Benzene, Methanol and Ethanol.
145. Flash Point: It is defined as the lowest temperature at which a liquid can form an ignitable mixture in air near the surface of the liquid. The lower the flash point, the easier it is to ignite the material.
146. Retest Period: Retest Period is the period of time during which the Raw Materials and Packing Materials can be considered to remain within specifications, and therefore acceptable for use in the manufacture of a given drug product, provided that it has been stored under defined conditions.
147. Expiry Date: The date at which a product is no longer potent or of therapeutic value, determined by The date for a drug estimated for its shelf life with proper storage in sealed containers away from harmful and variable factors like heat and humidity.
148. Sterilization: Sterilization is a term referring to a process that eliminates or kills all forms of life, including transmissible agents (such as fungi, bacteria, viruses, spore forms, etc.)
149. What are the types of Volumetric Titration?
2. Redox Titration:
3.Precipitation Titration:
4.Complexometric Titration:
150. Normal Phase:- In chromatography/HPLC uses a polar stationary phase and a non-polar mobile phase.
151. Reversed Phase:- In chromatography/HPLC uses a non-polar stationary phase and a polar mobile phase.
152. What are the 4 principles of chromatography?
Four separation techniques based on molecular characteristics and interaction type use mechanisms of ion exchange, surface adsorption, partition, and size exclusion.
153. What is the principle of TLC?
The principle of Thin Layer Chromatography (TLC) is adsorption, where a mixture is separated based on the different affinities of its components for the stationary and mobile phases.
154. What is Rf value in TLC?
The R𝑓 value in Thin Layer Chromatography (TLC) is the retention factor, which is the ratio of the distance a compound travels to the distance the solvent front travels.
157. What is Bulk Density?
Bulk density is the mass of a substance divided by the total volume it occupies, including the space between particles (voids). It is a measure of how much a material weighs per unit volume.
158. What is Tap Density?
Tap density is the ratio of a powder’s mass to its volume after the powder has been mechanically tapped to a more compact state.
159. What is Density?
Density is the measure of mass per unit volume.
Density = Mass/ Volume
159. What is Specific Gravity/ Relative Density?
The ratio of a substance’s density to the density of a reference substance (Water).
159. What is Weight per milliliter (wt/ml)?
Weight per milliliter (wt/ml) is a measure of density with units like grams per milliliter (𝑔/𝑚𝑙). Wt/ml tells absolute weight of particular volume.
160. What is the basic principle of KF?
The core of the method is the reaction where water (𝐻2𝑂) reacts with iodine (𝐼2) and sulfur dioxide (𝑆𝑂2) to form sulfuric acid (𝐻2𝑆𝑂4) and hydrogen iodide (𝐻𝐼). The excess iodine can be detected using a sensitive electrochemical method to mark the endpoint. It is water detection method in sample.
161. What is KF Reagent?
Karl Fischer reagent is a combination of iodine, sulfur dioxide, and water in a buffer solution (like imidazole or pyridine) and a solvent (like methanol). The overall reaction is;
162. Calculation of water content: Plug the values from the titration into the main formula:
% Water = (Volume of KF Reagent x KF Factor x 100) /Sample Weight (g)
163. What is Oxidation?
The loss of electrons or addition of Oxygen in substance.
e.g. C + O2 → CO2 (oxidation of carbon)
164. What is Reduction?
The gain of electrons or addition of Hydrogen in substance.
e.g. N2 + 3 H2 → 2NH3 ( reduction of nitrogen)
165. What are mineral Acids?
Sulfuric acid (𝐻2𝑆𝑂4), Hydrochloric acid (𝐻𝐶𝑙), and Nitric acid (𝐻𝑁𝑂3).
166. What is difference Between Organic and Inorganic Compounds?
Organic compounds always have a carbon atom, while most of the Inorganic compounds do not contain a carbon atom in them. Almost all organic compounds contain carbon-hydrogen or a simple C-H bond in them.
167. What is difference between Molecular weight and Equivalent Weight?
Molecular weight is the mass of one mole of a substance, a fixed value for a given compound.
Equivalent weight, is a variable value that depends on the substance’s role in a specific chemical reaction, calculated by dividing the molecular weight by its “n-factor” or valence. The n-factor represents the number of reactive units, such as protons or electrons, that a molecule can donate or accept.
168. Which electrode used in pH Meter?
A pH meter uses a glass electrode to measure the hydrogen ion concentration and a reference electrode (most commonly a silver-silver chloride electrode) to provide a stable, unchanging potential.
169. which solution used to store pH electrode?
A pH electrode should be stored in solution of potassium chloride (KCl), to keep the glass bulb hydrated and prevent it from drying out.
170. How to activate pH electrode?
Dip pH Electrode in a 1M Hydrochloric Acid (𝐻𝐶𝑙) solution for several hours/overnight.
171. What is the principle of UV Spectroscopy?
The Beer-Lambert law is the principle behind UV-Vis spectroscopy, which explains that light absorbance is directly proportional to both concentration (𝑐) and path length (𝑙).
172. What UV & Visible Range?
Ultraviolet (UV) region from 190 to 400 nm and the visible light region from 400 to 800 nm
173. What are the lamp/light source in UV Spectroscopy?
Deuterium lamp for UV light and a Halogen or Tungsten lamp for visible light as a source of light.
174. What is stray Light?
Stray light in UV-Vis spectroscopy is any light that reaches the detector which is not at the specific wavelength set by the monochromator, leading to significant errors in absorbance measurements.
175. What is Monochromatic light?
Light with a single wavelength or frequency.
176. What is the MOC of UV Cuvettes?
Cuvettes can be manufactured from a variety of materials such as plastic, glass, and quartz.
177. What is Tailing Factor?
Symmetry factor (S, also called “tailing factor”) is a coefficient that shows the degree of peak symmetry.
178. What is Theoretical Plates?
Theoretical plates (N) are a measure of the efficiency of a High-Performance Liquid Chromatography (HPLC) column.
179. What is Resolution in HPLC?
Resolution (𝑅𝑠) is a measure of how well two adjacent peaks are separated from each other in a chromatogram.
180. What is Capacitor Factor?
The capacity factor (also called the capacity ratio, (𝑘′) is a measure of how well a compound is retained by the stationary phase.
181. What is void volume?
Void volume (𝑉𝑚) in HPLC is the total volume of the mobile phase within a column, representing the space not occupied by the stationary phase.
182. What is dead volume?
Dead volume is a broader term for the total volume in the system where no separation occurs. Starting from Injector till detector.
183. What is the C8 and C18 HPLC column?
C18 column uses 18-carbon chains and C8 column uses shorter, 8-carbon chains.
184. Is C18 polar or non-polar?
C18 is nonpolar because it has a long hydrocarbon chain (18 carbons) bonded to a silica support.
185. Why RT Shift in HPLC?
Retention time (RT) shifts in HPLC are caused by changes in the mobile phase composition or flow rate, column temperature variations, and system issues like leaks or pump problems.
186. What is RRT?
RRT in HPLC stands for Relative Retention Time, a ratio that compares the retention time of an analyte to the retention time of a reference compound.
187. What is RRF?
RRF (Relative Response Factor) is a factor used for quantitative analysis of impurities, relating the detector response of a compound to a reference compound.
188. What is TLC Plate?
It is combination of the plate’s stationary phase, support material, and any additional features like a fluorescent indicator. Common stationary phases include silica gel and alumina, while common support materials are glass, aluminum, or plastic.
189. What is Principle of IR?
The principle of Infrared (IR) spectroscopy is that molecules absorb infrared radiation at specific frequencies that match the vibrational frequencies of their chemical bonds, such as stretching or bending.
190. What is IR Range?
The full IR spectroscopy range is typically 12,800–10 cm⁻¹
Mid IR-4,000–400 cm⁻¹
191. What Light Source/Lamp used in IR spectroscopy?
IR spectroscopy light sources include Globar (a silicon carbide rod), Nernst glowers, and halogen lamps.
192. How calibration is performed in IR spectroscopy?
Standard polystyrene film is used check the wavenumber accuracy and resolution, comparing the resulting spectrum to known reference peaks.
193. Why KBr is used in IR spectroscopy?
KBr is used because it is optically transparent in the IR range (4000-400 cm⁻¹), has a neutral salt that doesn’t absorb IR radiation.
194. What is the principle of Atomic absorption spectroscopy (AAS)?
The process involves atomizing the sample, exposing it to light of a specific wavelength from a hollow cathode lamp, and then measuring how much light is absorbed by the sample’s atoms as they make the transition from the ground state to an excited state.
195. What Light Source/Lamp used in AAS?
Hollow-cathode lamp (HCL).
196. What is the principle of Mass spectroscopy?
Mass spectrometry principle is to ionize a sample, separate the ions based on their mass-to-charge ratio (𝑚/𝑧), and then detect them to determine their relative abundance.
197. What Light Source/Lamp used in Mass spectroscopy(MS)?
Xenon lamps.
198. Which compound used in MS calibration?
Calibration is performed using specific calibration compounds (like perfluorotributylamine, sodium cesium iodide, or polyethers).
199. What is Gas chromatography-Mass spectrometry (GC-MS)?
The GC separates volatile components based on their chemical properties, while the MS acts as a detector to provide information on the mass-to-charge ratio of each separated component, allowing for identification and quantification even at trace levels.
200. What is Liquid chromatography-Mass spectrometry (LC-MS)?
An analytical technique that combines liquid chromatography (LC) to separate compounds in a mixture with mass spectrometry (MS) to identify and quantify them based on their mass-to-charge ratio.
201. What is Gas chromatography (GC)?
Gas chromatography (GC) is a common analytical technique used to separate, identify, and quantify volatile compounds in a mixture. It works by passing a mobile gas phase through a stationary phase (a column), which separates compounds based on their boiling points, polarity, and adsorption properties.
202. Which detector/Lamp used in GC?
A photoionization detector (PID) lamp or Flame Ionization Detector (FID) or Thermal conductivity detector (TCD).
203. What are the types of GC Columns?
The two main types of gas chromatography columns are packed columns and capillary columns. Packed columns are filled with a solid support material coated with a stationary phase and are used for larger sample volumes or specific applications like analyzing permanent gases. Capillary columns are narrow tubes with a stationary phase coated on the inner wall, providing higher resolution and faster analysis for more complex mixtures.
204. What is the difference between Polar and Non-Polar Molecules?
Polar molecules have an unequal sharing of electrons, creating partial positive and negative ends, while nonpolar molecules have an equal sharing, resulting in no distinct poles.
205. What is ICP-MS?
ICP-MS (Inductively Coupled Plasma Mass Spectrometry) is an analytical technique that uses a high-temperature plasma to ionize elements in a sample and a mass spectrometer to separate and detect these ions based on their mass-to-charge ratio.
206. Why is argon used in ICP-MS?
Argon gas is used in ICP-MS because it is an inert, chemically stable, and abundant gas that is ideal for creating a high-temperature plasma to ionize the sample.
207. Which detector is used in ICP-MS?
The most common detector used for ICP-MS is an electron multiplier (EM).
208. What is Dry Ice?
Dry ice is the solid form of carbon dioxide (CO2).
209. What is Thermogravimetric analysis (TGA)?
Thermogravimetric analysis (TGA) is a technique that measures the weight change of a material as a function of temperature or time.
It is used to study a material’s properties, such as its thermal stability, composition, purity, decomposition reactions, and absorbed moisture content, by heating a sample in a controlled atmosphere.
210. What is Gravimetric analysis ?
Gravimetric analysis is a quantitative chemical technique that determines the amount of a substance by measuring its mass.
The four types of gravimetric analysis are precipitation, volatilization, electrogravimetry, and thermogravimetry.
211. What is the principle of colorimetry?
The absorbance of light by a colored solution is directly proportional to the concentration of the absorbing substance and the path length of the light through the solution.
212. What is principle of Autoclave?
An autoclave is a pressure chamber used to sterilize instruments and materials through moist heat, using high-pressure steam to kill microorganisms like bacteria, viruses, and spores.
213. What is Biological Indicator?
A biological indicator is a test system containing highly resistant microorganisms, typically bacterial spores, used to monitor and ensure the effectiveness of sterilization processes like steam, gas, or dry heat.
214. What is the most commonly used biological indicator?
270. Which lamp is used in a refractometer?
sodium vapor lamp.
271. What is the principle of melting point?
The principle of melting point is the temperature at which a substance transitions from a solid to a liquid state, where the solid and liquid phases are in equilibrium.
272. What is viscosity?
Viscosity is a fluid’s resistance to flow.
𝜂= 𝐹____________
𝐴×𝑑𝑣/𝑑𝑥
where;
𝜂(eta)= is the dynamic viscosity,
𝐹= is the shearing force,
 𝐹= is the shearing force,
𝐹= is the shearing force,𝐴= is the area, and
𝑑𝑣/𝑑𝑥= is the velocity gradient
273. What is potentiometric titration?
Potentiometric titration is an electroanalytical technique that measures the change in electrical potential between two electrodes to determine the equivalence point of a titration.
274. How end point is detected in Potentiometric Titrations?
The endpoint of the titration is reached when there is a sharp and sudden change in the measured potential, which occurs when the reaction is complete. At the equivalence point, the concentration of the ions involved in the reaction changes dramatically, causing a steep and sudden change in the potential.
275. BCS Classification of Drug Substance:
Class I – High Solubility, High Permeability
Class II – High Permeability, Low Solubility
Class III -High Solubility, Low Permeability
Class IV – Low Solubility Low Permeability
276. What is BMR?
BMR: Batch Manufacturing Record. A document stating the materials used and the operation carried out during the processing of a given batch including details of in process control. It should be based on the master formula and be complied as the manufacturing operation process. BMR contents product name, strength, Batch no, Batch size, Quantity, Quantity of raw material, code no, QC reference no, Calculation on potency, manufacturing procedure, temperature, humidity, time, Yield of different stage etc.
277. What is BPR?
BPR: Batch Packaging Record. A document stating the bulk product and packaging material used and the process carried out during the packaging of a given batch with details in process control. It should be based on the master formula & packaging instruction and be complied during the packaging operation. BPR contents Product name, strength, Batch no, Batch size, Pack size, MRP, Mfg. date, Exp. Date, Outer box / Shipping carton size etc.
278. What is Reprocessing?
The reworking of all or part of batch of product of an unacceptable quality from a defined step of production in order its quality may be rendered acceptable by one or more additional operations. Reprocessing or reworking may be required for many reasons the most common causes are potency adjustments, failure to meet physical parameters, non uniform blending etc.
279. What is Class-100 in Clean room?
The area in which particle count not to exceed more than 100 particles per cubic feet of a size 0.5 μm and large size.
280. What is Non-Conformity?
A deficiency in a characteristic, product specification, process parameter, record, or procedure that renders the quality of a product unacceptable, indeterminate, or not according to specified requirements.
281. What is FAT & SAT?
FAT stands for Factory Acceptance Test, a series of tests performed at the equipment manufacturer’s site to verify that a new piece of equipment works according to the agreed-upon design specifications and client requirements before it is shipped.
“SAT” stands for Site Acceptance Test, a crucial process that verifies equipment is functioning correctly at the user’s site after installation. This involves testing the equipment in its intended environment to ensure it meets all requirements, integrates properly, and is ready for operational use.
282. What is Impact Assessment?
It formal process used to identify systems and the components of those systems that have a direct impact on Product Quality.
283. What is Gap analysis?
Gap analysis is a process of comparing current practices against desired standards, regulations, or goals to identify discrepancies that need to be addressed.
284. What is GAMP5 (Good Automated Manufacturing Practice)?
A risk based approach to compliant GxP computerized systems provides a frame work for the risk-based approach to computer system validation where a system is evaluated and assigned to a predefined category based on its intended use and complexity.
285. What is Residual Solvents?
A residual solvent is an organic chemical used in the manufacturing of a product, such as a pharmaceutical or food item, that is not completely removed by the end of the process.
286. What are the Classes of Residual Solvents?
- Class 1: Solvents to be avoided due to high toxicity (Benzene, Carbon tetrachloride, 1,2-dichloroethane,).
- Class 2: Solvents that should be limited. (Acetonitrile, Chloroform,Methanol)
- Class 3: Solvents with low toxic potential. (Acetic Acid, Ethanol,IPA)
287. What is Hold time study?
Hold time is the established period for a material, intermediate, or bulk product can be held at a specific stage before further processing without compromising its quality, safety, or efficacy.
288. What is Heavy Metals?
A metallic chemical elements with high density that can be toxic at low concentrations, such as lead, mercury, cadmium, and arsenic.
They can accumulate in the body and cause poisoning, leading to various health problems.
289. What is metal detection test?
A tablet metal detector is a specialized machine used in the pharmaceutical industry to detect and remove tiny metal particles from tablets and capsules before they are packaged.
290. What is Blending?
Blending in pharmacy is the process of thoroughly mixing pharmaceutical powders, including active pharmaceutical ingredients (APIs) and excipients, to create a uniform and homogenous mixture for a final dosage form.
291. What is Granulation?
Granulation in pharmaceuticals is a process of agglomerating fine powders into larger, free-flowing granules to improve flowability, compressibility, and uniformity for producing tablets and capsules.
292. What is wet Granulation & Dry Granulation?
Wet granulation, which uses a liquid binder, and dry granulation which uses mechanical force without a liquid binder.
293. What is Compression?
The process of forming solid dosage forms, like tablets, by applying pressure to a mixture of powdered active pharmaceutical ingredients and excipients.
294. What is Tablet tooling?
The dies and punches and their setup on compression machine is called tooling , it is classified as B and D tooling .
295. What is Passivation?
A crucial chemical process that removes free iron from stainless steel surfaces, creating a thin, protective chromium oxide layer that enhances corrosion resistance.
296. What is tablet Coating?
It is the process of applying a thin, dry, outer layer of material (typically a polymer) to the surface of a solid dosage form such as a tablet, capsule, granule, or pellet.
297. What is sampling Thief?
It is a tool used to collect samples from powders and granules in drums, bags, or other containers.
It is a tubular device, often made of stainless steel, that is inserted into the container to capture a sample from a specific depth, providing a representative and accurate sample for quality control testing.
298. What is dispensing?
The dispensing is the process of accurately measuring and transferring specific quantities of Raw Materials for production.
299. What is Sifting & Milling?
Sifting is a process of passing powders or granules through a sieve to ensure uniform particle size, remove impurities, while Milling reduces the particle size of solids by breaking up large lumps.
300. What is Lubrication?
Lubrication is the process of adding lubricants to tablet formulations to reduce friction during manufacturing. Lubricants prevent powders from sticking to equipment, improve the flow of the powder blend.
301. What is Sieve Analysis?
Sieve analysis is a test to determine the particle size distribution of a powder by passing it through a series of sieves with progressively smaller openings.
302. What line Clearance?
Line clearance is a critical, systematic process that ensures a manufacturing or packaging line is completely free of residues, materials, and documentation from a previous product before a new batch or product can begin. It is a mandatory step to prevent cross-contamination, mix-ups, and errors, thereby protecting product quality and patient safety.
303. What is CIP & SIP?
CIP (Cleaning-in-Place) and SIP (Sterilization-in-Place) are automated processes in pharmaceuticals for cleaning and sterilizing equipment without disassembly. CIP uses chemicals, water, and heat to remove residues, while SIP uses clean steam to destroy all microorganisms.
304. What is APQR?
APQR, or Annual Product Quality Review, is a mandatory yearly evaluation of a pharmaceutical product’s quality to ensure it meets specifications and regulatory standards. This process involves analyzing manufacturing, quality control, and any issues or changes that occurred during the year to identify trends, implement corrective actions, and maintain product integrity.
305. What is DMF?
A Drug Master File (DMF) in pharma is a confidential submission to regulatory agencies like the USFDA/MHRA that provides detailed information about the manufacturing, processing, packaging, and storage of a drug substance, ingredient, or product.
306. What is Environment monitoring?
Environment monitoring is a systematic process of assessing the cleanliness and quality of a manufacturing environment to ensure pharmaceutical products are safe. It involves regularly sampling air, surfaces, and personnel to detect and control microbial and particulate contamination.
306. What is Stress Testing?
stress testing, also known as forced degradation studies, is the process of deliberately subjecting new drug substances and products to extreme chemical and environmental conditions to evaluate their stability and identify potential degradation pathways and products.
307. What is Stability Indicating testing Method?
A analytical procedure that can accurately measure chemicals and physical changes in product over the time. For example- content of active pharmaceutical ingredient (API) while distinguishing it from any degradation products, impurities, and excipients.
308. What is Physical Parameters?
The measurable characteristics of a drug product, like tablets or capsules, that are essential for quality control, safety, and effectiveness. Examples include color, hardness, thickness, weight variation, friability, and disintegration time, as well as properties of the raw materials like particle size and moisture content.
309. What is pilot Batches & scale up batches?
A pilot batch is an intermediate-sized production run made in a pilot plant to test and refine a manufacturing process developed at the lab scale. Scale-up is the overall process of increasing production from the lab, through the pilot stage, to commercial large scale.
310. What is URS?
User Requirements Specification (URS) describes what users require from the System. User requirement specifications are written early in the validation process, typically before the system is created.
311. What is QbD?
QbD stands for Quality by Design, a systematic approach to drug development that builds quality into a product from the beginning through a thorough understanding of its development and manufacturing processes.
It is systematically build quality into a product and its manufacturing process from the start, based on scientific understanding and risk management.
312. What are the 7 QC tools?
The 7 Quality Control (QC) tools are: a check sheet, a fishbone (cause-and-effect) diagram, a histogram, a Pareto chart, a scatter diagram, a control chart, and stratification (or sometimes a flowchart).
313. What is CBE-30 filing?
CBE-30 stands for “Changes Being Effected in 30 Days”. It is a type of regulatory submission to the U.S. Food and Drug Administration (USFDA) for making moderate changes to an approved drug product after it’s already on the market.
314. What is the recommended bio burden limits of purified water & WFI?
Purified water has a recommended bio burden limit of 100 CFU/mL, and water for injection (WFI) has a recommended bio burden limit of 10 CFU/100 ml..
315. What is the pass/fail criterion for disintegration test (DT)?
If one or two tablets/capsules fails to disintegrate completely, repeat the test on another 12 additional dosage units. The requirement is meet if not fewer than 16 out of 18 tablets/capsules tested are disintegrated completely.
316. What is the pass/fail criterion for Dissolution Test?
S1 tests 6 units, requiring each to be ≥Q+5%. If S1 fails, S2 proceeds with 6 additional units (total 12), allowing some units below the S1 limit but not below Q-15%. If S2 fails, S3 tests 12 more units (total 24), requiring the average to be ≥Q with specific limits and not more than 2 units below Q-25%.
317. What is the limit of weight variation?
Weight Variation Limits USP Limits –
130 mg or less: ±10%
130 mg to 324 mg: ±7.5%
More than 324 mg: ±5%
318. What is the Acceptance criteria for Content Uniformity (CU)?
- L1: The maximum allowed Acceptance Value 15.0% for 10 Units.
- L2: The maximum allowed Acceptance Value 25.0% for 30 Units.
319. What is Carcinogen?
A carcinogen is any substance, organism, or agent that can cause cancer by damaging a cell’s DNA.
320. What is Oncology products?
Oncology products are pharmaceuticals and other treatments specifically designed to diagnose, treat, and manage cancer.
321. What is Hormonal products?
Hormonal products are medications containing active pharmaceutical ingredients that mimic or regulate the body’s own hormones to treat a variety of conditions.
322. What is Cytotoxic products?
Cytotoxic products are substances that are toxic to cells, meaning they kill or inhibit cell growth. Most commonly used for chemotherapy drugs used to kill cancer cells.
323. What is Antibiotic products?
Antibiotic products are a class of powerful medications designed to treat or prevent infections caused by bacteria. They work by either killing the bacteria (bactericidal) or preventing them from growing and multiplying.
324. How to perform qualification of pharmaceutical water system?
In 3-Phases.
Phase-1; Test period of 2-4 weeks should be spent for monitoring the system intensively. Under take chemical & microbiological testing.
Phase-2; Further Test period of 2-4 weeks should be spent for monitoring the system intensively. Under take chemical & microbiological testing.Water can be used for manufacturing process in this phase.
Phase-3; Phase 3 runs for one year after satisfactory completion of phase-2. Under take chemical & microbiological testing. Ensure that seasonal variations are evaluated. Water can be used for manufacturing process in this phase.
325. What is preventive maintenance?
It is periodic inspection and minor repairs of equipment as per define schedule . This Enables smooth operation and long life of the equipment. It also avoids major breakdown of the equipment during manufacturing of the product.
326. What is Batch ?
A specific quantity of material produced in a process or series of processes so that it is expected to be homogeneous within specified limits.
327. What is quarantine?
The status of materials isolated physically or by other effective means pending a decision on their subsequent approval or rejection.
328. What is the difference between instrument & equipment?
Instrument: is a more specific device used for precise measurement or analysis. Device takes a physical measurement and displays a value. (HPLC,FTIR,pH Meter)
Equipment: is a broad term for a set of tools or machines used to carry out particular process or activity. Assembled to perform a mechanical process. (FBD, RMG, Blender).
329. What is “Reference standard” and “Working standard”?
Reference Standard (RS): A substance that has been shown by an extensive set of analytical tests to be authentic material that should be of high purity. This standard may be obtained from an officially recognized source or may be prepared by independent synthesis or by further purification of existing production material.
Working Standard (WS): A substance that established quality and purity by comparing with reference standard. WS used for routine laboratory analysis. its cost saving.
330. Describe the categories of the market complaints?
Market complaints are categorized into three types and are as follows;
Critical: Complaints related to suspected contamination, adulteration and mislabeling.
Major: Complaints related to the product not meeting its pre-determined critical specifications and damage to primary packaging.
Minor: Complaints related to the product not meeting non-critical quality attributes, or damage to secondary packaging or shortages etc.
331. What are the types of Non-Compliances?
Non-compliances shall be categorized as follows:
Critical: Those findings that warrant stoppage of any further operations in the facility until the corrective actions have been completed.
Major: Those findings that require immediate corrective action plan and compliance although operations can be continued.
Minor: Those findings that require corrective action plan as agreed between the Auditee and Auditor.
332. What do you mean by “worst case study”?
A study which is carried out by deliberately exceeding upper and lower processing limit to check the impact on product or activity or area which poses the greatest chance of product or process failure when compared to ideal conditions.
333. What is the difference between controlled copy and uncontrolled copy?
Controlled copy: A controlled copy is a formal copy of the latest, correct issue of a document; an identified issue of a document to an individual or location of record. The controlled copy is officially tracked, updated & destroyed to assure that it is current.
Uncontrolled copy: An informal copy of a document for which no attempt is made to update if after distribution; the document is marked “uncontrolled” and the user determines if the document is active prior to use.
334. What are the system suitability parameters?
Tailing factor, Theoretical plates, Relative Standard deviation (RSD), Resolution, Retention time (RT), Relative retention time (RRT), Capacity factor.
335. What is reconciliation?
A stock taking between the amount of product or material theoretically required & actually used.
336. What is the QMS?
It is the quality management system to direct and control an organization with regard to quality.
337. What is retention/Control sample & why retention sample is preserved?
A part of the sample which is representative of the released batch of a finished product preserved beyond its shelf life.
It is preserved for future reference /investigation/ reanalysis in cases of market complaints.
338. What is “PRODUCT RECALL”?
A removal of distributed product from market for the reasons relating to deficiencies in quality, safety or efficacy, including labeling considered to be in violation of the laws.
Class I Recall: Notification and acknowledgement of receipt of recall notification within 24hrs.
Class II Recalls: Notification and acknowledgement of receipt of recall notification within 48 hours.
Class III Recalls: Notification and acknowledgement of receipt of recall notification within 5 days.
339. What is the difference between Contamination, Cross Contamination and Mix-Up?
Contamination- The presence of any other substance other than product manufacturing formula. It ma be chemical, physical or microbial.
Cross Contamination- Transfer of one product or material with another product.
Mix-Up- A mix-up is the unintentional substitution or incorrect labeling of products, materials, or documents, where the wrong item is packaged with the wrong label or in the wrong place.
340. What is Pass Box?
It is an enclosed cabinet with two interlocked doors used to transfer materials between areas without compromising the cleanroom integrity.
341. What is static and dynamic pass box?
A static pass box is a passive transfer chamber used between two areas of the same cleanliness level, with no internal air handling or filtration.
A dynamic pass box is an active system with built-in blowers and HEPA filters, used to transfer materials from an uncontrolled to a controlled cleanroom environment, or between areas of different cleanliness levels.
342. Why to perform tap density & bulk density?
It is crucial for assessing powder flowability, compressibility, and packing, ensuring consistent tablet/capsule quality, uniform dosing, and efficient manufacturing by reducing air voids, guiding formulation design (like granulation), and meeting strict USP/EP regulatory standards
It help select appropriate equipment (hoppers, fillers), control tablet hardness/dissolution.
343. What is role Particle size?
Particle size in pharma is crucial because it controls a drug’s bioavailability, affecting how fast it dissolves and gets absorbed, and also impacts manufacturing (flow, blending, compression), stability, and specific delivery (inhalation, IVs).
344. What is Genotoxic impurities?
Genotoxic impurities are chemicals that can damage genetic material (DNA) and potentially cause mutations, which may lead to cancer.
345. What is Hygroscopic Material?
In pharma, a hygroscopic material is a substance (drug or excipient) that readily absorbs moisture (water) from the surrounding environment (air), which significantly affects its physical stability (clumping, flow) and chemical stability (degradation, reduced efficacy), necessitating strict humidity control during manufacturing, storage, and packaging to maintain product quality and shelf life.
346. What is Hydrolysis and its impact?
Hydrolysis is the chemical breakdown of a drug by reaction with water, which can occur both as a degradation process and as a method for activating a prodrug. It is a common issue in drug formulation and storage, as it can decrease the concentration of the active pharmaceutical ingredient (API) and create new potentially inactive or toxic substances (Impurity).
347. What is Clinical Trials?
Clinical trials are research studies that test the safety and effectiveness of new drugs in treatments in human volunteers.
The clinical trial process involves several phases to test new drugs and treatments for safety and effectiveness. The process begins with preclinical research, followed by four phases in human subjects: Phase I (safety and dosage in a small group), Phase II (effectiveness and side effects in a larger group), Phase III (efficacy compared to standard treatments in a large group), and Phase IV (long-term safety after approval).
348. What is role binder/biding agent?
A binder (or binding agent) is a crucial excipient that acts like an “glue,” holding powders, fillers, and the Active Pharmaceutical Ingredient (API) together to form a strong, cohesive solid dosage form like a tablet or granule, preventing crumbling during manufacturing and use, improving flow, and sometimes controlling drug release.
E.g. Povidone (PVP), polyethylene glycol (PEG), copovidone, Hydroxypropyl methylcellulose (HPMC).
349. What is role lubricating agent?
Lubricating agents are essential for tablet and capsule manufacturing to reduce friction, prevent sticking, and improve powder flow. Lubricants ensure smooth powder flow, protect tablet-making equipment by reducing wear, and help with the ejection of tablets from the die walls.
E.g. Magnesium stearate, Talc, Colloidal silicon dioxide.
350. What is role disintegrating agent?
A disintegrating agent (disintegrant) is a crucial pharmaceutical excipient added to solid dosage forms (like tablets and capsules) to facilitate their breakup into smaller particles when they come into contact with bodily fluids, such as in the gastrointestinal tract.
Disintegrants ensure that the active pharmaceutical ingredient (API) is released quickly and efficiently so it can dissolve and be absorbed into the bloodstream, producing the desired therapeutic effect.
E.g. Starches, Microcrystalline cellulose (MCC).
351. Why Coating is required for Tablets?
Many active pharmaceutical ingredients (APIs) are sensitive to environmental factors such as moisture, light, and oxygen. A coating acts as a protective barrier, which enhances the stability and extends the shelf life of the medication.
Also, Coating of tablets can mask unpleasant Taste and odor of drug.
352. What is Immediate release, Modified release and Extended release tablets?
Immediate Release (IR): This is the conventional dosage form, designed to dissolve quickly and release the medication all at once for fast absorption and rapid therapeutic effect.
Modified Release (MR):This is the modified dosage form which alters the rate, time, or location of the release of tablet based on requirements.
Extended Release (ER): This specific type of modified release is engineered to deliver the drug slowly over an extended period, typically 12 to 24 hours. The goal is to provide a prolonged therapeutic effect, reduce dosing frequency, and maintain consistent drug levels in the bloodstream to minimize side effects.
353. What is chemical compatibility chart?
A chemical compatibility chart is a reference guide that rates how different chemicals react with each others, helping users to choose safe combinations for handling, storage, or equipment construction to prevent dangerous reactions, corrosion, or material failure.
354. What is HAZOP study?
A HAZOP study, or Hazard and Operability Study, is a systematic and structured risk assessment method used to identify potential hazards and operational problems in a complex process or system
355. what is HACCP study?
Hazard Analysis and Critical Control Points.
Identify and assess potential hazards that could be present in the process. Identify the points in the process where hazards can be prevented, eliminated, or reduced to an acceptable level.
356. What is FTA (Fault Tree Analysis)?
It is hazard analysis technique, Top-down Approach, event-focused; starts with an undesired event (e.g., explosion) and traces all contributing causes (gates).
357. What is FMEA (Failure Modes and Effects Analysis)? Analyzes potential failure modes of components, their causes, and effects.
358. What is HIRA?
HIRA stands for Hazard Identification and Risk Assessment, a systematic process for identifying potential hazards in a workplace or process , evaluating the associated risks, and implementing control measures to prevent accidents and ensure safety.
359. What is OSHA?
OSHA (Occupational Safety and Health Administration) is a U.S. federal agency that ensures safe and healthy working conditions for employees by setting and enforcing safety standards, providing training, and offering assistance.
360. What is “Near Miss” in safety?
A near miss in safety is an unplanned event or identification of hazard that could have resulted in injury, illness, or property damage, but did not. These events are crucial warning signs of potential hazards and provide valuable opportunities to implement preventative measures before a real accident occurs.
361. What is PPE in safety?
Personal Protective Equipment (PPE) is a barrier between a person and hazards in the workplace, designed to protect the wearer from serious injury or illness. It includes a wide range of gear, such as hard hats, safety goggles, gloves, helmets, Shoes, Face shield, safety suit, respirators, and high-visibility clothing, and is used to shield against hazards like physical, chemical, electrical, and biological risks.
362. What is Environmental Monitoring?
Environmental Monitoring (EM) in the pharmaceutical industry is a systematic program of sampling and testing the manufacturing environment to detect and control microbial and particulate contamination.
363. What is Nitrosamine Impurities?
Nitrosamine impurities in pharmaceuticals are trace chemical contaminants that are classified as probable human carcinogens (cancer-causing agents).
364. How many types of impurities are there in pharma products?
There are three main types of impurities: organic, inorganic, and residual solvents.
Organic impurities: These are by-products, intermediates, or degradation products of the main substance. They can originate from starting materials, reagents, or other organic chemicals used in the manufacturing process.
Inorganic impurities: These are typically inorganic compounds like reagents, ligands, catalysts, heavy metals, or inorganic salts that are carried over from the manufacturing process.
Residual solvents: These are the liquids used during the manufacturing process that are not completely removed from the final product.
365. What is difference between Assay, Purity and Potency?
Assay measures the total amount of API in drug product.
Purity checks impurities present in product.
Potency quantifies its biological effectiveness or strength of the drug.
366. What is the difference between purity on Dried basis and purity Anhydrous Basis?
Purity on a dried basis removes moisture and volatile solvents through drying, reflecting the active substance in the dry residue.
Purity on an anhydrous basis is a calculated value removing only water, assuming no other volatiles, giving a truer measure of the absolute active compound by stripping water of hydration.
367. Why KBr used in FTIR/IR spectroscopy?
Potassium bromide (𝐾𝐵𝑟) is used in IR spectroscopy because it is optically transparent to IR radiation, meaning it does not absorb the infrared light being used for analysis in the relevant wavenumber range (4000−400 𝑐𝑚−1).
368. What is the difference between amorphous and crystalline materials?
Crystalline materials have a highly ordered, repeating atomic structure crystals.
Amorphous materials have a disordered, random arrangement of atoms like powder.
369. Why caffeine used in HPLC calibration?
Caffeine is used for HPLC calibration because it is readily available, stable, highly soluble, and produces a sharp, symmetrical peak with a well-defined retention time. It also has strong UV absorbance, making it easily detectable with standard UV detectors.
370. What is the difference between Isocratic and Gradient elution in HPLC?
Isocratic elution is fixed composition of mobile phase throughout entire run time,
Gradient elution varies the mobile phase composition over time.
371. What is sink condition in dissolution testing?
Sink condition can be determined by finding the saturation solubility of drug in the media and multiplying the media volume by 3.
Sink Condition = 3 x Saturation Volume.
372. What is the temperature used in dissolution testing?
Temperature for dissolution testing around 37±0.5°C, mimicking body temperature, because temperature significantly affects drug solubility, dissolution rates, and overall drug release in body.
373. What is the temperature limit for DT?
The standard temperature limit for a disintegration (DT) test is 37±1∘𝐶. Ensures physiological conditions of the human body.
374. What is Mesh Size?
The “number of openings per inch”
375. What are specific temperature ranges for pharmaceutical storage?
a. Controlled Room Temperature (CRT): 20°C–25°C with allowed excursions between 15°C–30°C.
b. Cool: 8°C–15°C
c. Refrigerator: 2°C–8°C
d. Freezer: -25°C to -10°C
e. Warm: 30°C–40°C
f. Excessive Heat: Above 40°C
376. Why pH meter is calibrated before use or daily?
A pH meter is calibrated before use to ensure accurate and reliable measurements, as the pH electrode’s properties change over time due to aging and drift, affecting its readings. Calibration corrects for these changes by comparing the meter’s reading to standard buffer solutions with known pH values and adjusting the meter’s output to match the standard.
377. Why analytical balance is calibrated before use or daily?
Analytical balances must be calibrated before use to ensure accuracy and reliability by correcting for drift from environmental changes (temp, humidity, gravity) and wear, preventing errors in critical measurements that affect experiments, quality control, and research outcomes. Without calibration, results can be inconsistent, leading to faulty data, wasted materials, and costly rework or recalls.
378. Why KF water factor is calibrated before use or daily?
The KF reagents degrade or change concentration, altering their reactivity with water.
The KF reagent’s water-equivalency (how much water 1 mL of reagent can titrate) changes over time and with use due to factors like reagent age, environmental humidity, temperature, and component wear.
380. What is the Maximum Weight can be weighted on any weighing balance?
In general, maximum 80% weight can be weighed on balance of its capacity.
E.g. 80kg can be weighed on 100kg capacity balance.
381. What is lamp hours in HPLC?
In HPLC, “lamp hours” refer to the usage time of the UV light source (usually a Deuterium lamp), typically guaranteed for around 2,000 hours.
382. Why Lamp Energy is important in HPLC?
Diminished lamp energy can result in a lower-than-expected peak response, potentially affecting the ability to accurately quantify compounds.
383. Which Lamp is used in HPLC?
A Deuterium lamp is the type of lamp most commonly used in HPLC to cover Ultraviolet (UV) range.
A Tungsten lamp used to cover the visible range.
384. What is Validation Master Plan?
A validation master plan (VMP) is a high-level document in the pharmaceutical industry that outlines the overall strategy for validating a company’s facilities, equipment, processes, and systems to ensure they consistently meet quality standards.
385. What is Pharmacopoeia?
A pharmacopoeia is an official, authoritative book setting quality standards (identity, purity, strength) for medicines, drugs, and related substances, published by a government or medical body.
USP (United States Pharmacopeia): Standards for the U.S
BP (British Pharmacopoeia): Standards for the UK
EP (European Pharmacopoeia): Standards for Europe
IP (Indian Pharmacopoeia): Standards for India.
386. What is difference between Expiry, Shelf life, Retest period, retest date?
Shelf Life: The period a product remains within its quality specifications, provided it’s stored correctly.
Expiry Date: The final date on a finished product (like a pill) after which it’s guaranteed unsafe/ineffective.
Retest Period: The timeframe for a drug substance or raw material (API) during which it’s expected to stay good and shall be retested.
Retest Date: The specific date after the retest period when the material must be re-tested, if it passes, it gets a new retest date and can keep being used.
387. What is FIFO and LIFO?
FIFO (First-In, First-Out) and LIFO (Last-In, First-Out) are methods for inventory management.
388. What is Impurity Threshold?
An impurity threshold limit is a defined concentration level in pharmaceuticals (or chemicals) that dictates required actions, like reporting, identifying the impurity’s structure, or providing toxicological data (qualification), based on the drug’s maximum daily dose (MDD) and international guidelines (ICH).
389. What is TOC?
A measurement of the Total Organic Carbon (TOC) in a sample, typically water.
390. What is Peak Purity?
Peak purity is a measure of a peak’s spectral homogeneity, checking if it contains just one compound or multiple ones (co-eluting impurities) by comparing UV/Vis spectra across the entire peak.It’s assessed using Photodiode Array (PDA) detectors and software, ensuring reliable quantification and identification.
391. What is sensitivity and selectivity in HPLC?
Sensitivity is a system’s ability to detect and amplify weak signals, while selectivity is its ability to distinguish and accept a desired signal while rejecting others. Sensitivity is about picking up a weak signal, and selectivity is about not picking up other, unwanted signals.
392. What is Design Space in QbD?
In Quality by Design (QbD), the design space is the multidimensional combination of input variables and process parameters that have been proven to provide assurance of quality for a product.This space is established through experimentation and risk assessments and, once approved by regulatory bodies, allows for changes within its boundaries without requiring resubmission.
393. What is Glassware Calibration?
Calibration of glassware is the process of verifying the accuracy of a volumetric instrument like Flask, pipette by comparing its measurements against a known standard, most commonly by weighing a precise volume of water.
394. What is difference between class A and class B glassware?
Lab glassware classes (A, B, ) primarily define accuracy/tolerance for volumetric measurements, with Class A being most precise (borosilicate glass, lower tolerance, for critical work) and Class B less precise (soda-lime, higher tolerance, for general use).
395. What are the common test performed in pharma?
Description, Identification, Assay, DT, Dissolution, Impurities. Related Substance, Friability, Hardness, Average Weight, Content Uniformity, Microbial Limits, pH, Viscosity, sterility.
396. What is ALCOA & ALCOA++?
ALCOA is a data integrity standard (Attributable, Legible, Contemporaneous, Original, Accurate) used in regulated industries, expanded to ALCOA+ by adding Complete, Consistent, Enduring, and Available, and further evolved into ALCOA++ by adding Traceable.
397. What is 21 CFR?
21 CFR is Title 21 of the U.S. Code of Federal Regulations, which contains rules and regulations governing food and drugs, including those from the Food and Drug Administration (FDA).
21 CFR Part 11, address electronic records and signatures, while others like 21 CFR Part 210 and 211 cover current Good Manufacturing Practices (cGMP) for drugs.
398. What are the regulatory authorities in pharma?
Pharmaceutical regulatory authorities which oversee drug safety, efficacy, and quality with key national bodies like the US FDA (USA), MHRA (UK), and CDSCO (India), TGA (Australia), ANVISA (Brazil) alongside global entities such as the WHO and ICH (International Council for Harmonisation).
399. What is Nitrogen purging in pharma?
Nitrogen purging in pharma is using inert nitrogen gas to displace harmful oxygen and moisture from equipment (reactors, tanks, pipelines) and final product packaging to prevent degradation, maintain stability, ensure sterility, and reduce explosion risks, thereby extending shelf life and guaranteeing product quality and safety.
400. What is Root Cause Analysis (RCA)?
RCA stands for Root Cause Analysis, a systematic, data-driven process to find the fundamental reasons behind quality failures, deviations, or problems (like a bad batch) to prevent them from recurring, ensuring patient safety, product integrity, and regulatory compliance.
401. What is Placebo?
The inactive ingredients (excipients) in Drug product called placebo.
402. What is Placebo effect?
The “placebo effect” is the psychological phenomenon where a person can experience a real improvement in their symptoms simply because they believe they are receiving an effective treatment.
403. What is AQL (Acceptable Quality Limit)?
AQL (Acceptable Quality Limit) is a statistical method used in quality control to determine the maximum number of defective items allowed in a batch to be accepted.
404. What is 1% solution?
A 1% solution contains one unit of solute per 100 total units of solution, most commonly interpreted as 1 gram of solute dissolved in a final volume of 100 milliliters of solution.
405. What is Turbid solution?
A turbid solution is cloudy or hazy because it contains suspended particles that are not fully dissolved. These particles scatter light, which causes the lack of transparency.
406. What is Opacity?
Opacity is the quality of being difficult to see through, meaning a substance blocks light (or other radiation) rather than letting it pass, appearing solid, unclear, or obscure. Opaque is opposite of transparency.
407. What is FLD Detectors?
An FLD detector, or Fluorescence Detector, is a highly sensitive scientific instrument used in chromatography (like HPLC) to detect and measure compounds that emit light (fluoresce) after being excited by a specific wavelength of light.
408. What is the difference between HPLC and UPLC?
UPLC is an advanced version of HPLC that uses smaller particles and higher pressures, resulting in faster analysis times, higher resolution, and greater sensitivity compared to the more traditional HPLC.
409. Why are C8 and C18 columns used in HPLC?
The choice between C18 and C8 columns depends on the specific separation requirements of the compounds being analyzed. C18 columns are typically more hydrophobic and offer stronger retention for nonpolar compounds, while C8 columns offer intermediate polarity and are suitable for separating moderately polar compounds.
410. What is C18 HPLC Column?
C18 refers to a specific type of stationary phase used in HPLC columns. It stands for Octadecyl, which indicates a chain of eighteen carbon atoms.
“C18” refers to an octadecyl group, which is a straight 18-carbon Hydrocarbon chain bonded to silica particles.
411. What is the pH range of C18 columns?
The pH range for a C18 column can vary, but it generally falls between pH 2 and 10, with some high-performance columns extending this range from pH 1 to 12.
412. What is a L1 & L7 HPLC column as per USP?
L1 column is an Octadecylsilane (C18) bonded phase on silica, while an L7 column is an Octylsilane (C8) bonded phase.
413. What is Ion Pairing reagent in HPLC?
An Ion Pairing (IP) reagent in HPLC is an additive put into the mobile phase to help separate ionic or highly polar compounds in reverse phase.
414. What is surfactant in dissolution?
A surfactant (surface-active agent) is a substance added to the dissolution medium (like water) to help poorly water-soluble drugs dissolve faster and more completely.
415. What is Photostability?
Photostability is the ability of a material to resist physical and chemical changes caused by exposure to light.
416. White is light sensitive drug product?
Medications/Materials that degrade, discolor, or form harmful impurities when exposed to light (especially UV/blue light).
417. How to protect light sensitive drug product/Materials?
By using controlled lighting (like brown light) during manufacturing, storage, and dispensing and Packing final product in amber color glass & opaque containers.
418. What is difference between the drug substance and drug product?
The drug substance (or API) is the pure, active ingredient providing the therapeutic effect, while the drug product is the finished, marketable form (like a tablet or capsule) that contains the drug substance mixed with inactive ingredients (excipients) and is ready for patient use, including its packaging.
419. What are the types of Fire & fire extinguisher?
Water: Primarily used for Class A fires (wood, paper, cloth).
Foam: Effective on Class A and Class B fires (flammable liquids like gasoline and paint) by smothering the flames.
Dry Chemical: Often labeled as “ABC,” these are multipurpose extinguishers suitable for Class A, B, and C (electrical) fires.
CO₂ (Carbon Dioxide): Ideal for Class B and Class C electrical fires because they displace oxygen and leave no residue, though they have a short range.
Wet Chemical: Specifically designed for Class K (or F) fires involving cooking oils and fats. They cool the fire and form a barrier to prevent re-ignition.
Dry Powder: These are specialist extinguishers for Class D fires involving combustible metals like magnesium, aluminum, or potassium.
420. What is Antidote?
An antidote is a medicine that counteracts against poison or the reduce harmful effects of a substance.
421. What are the color codes for gas cylinders?
Red Shoulder: Flammable (e.g., Hydrogen, Acetylene).
Yellow Shoulder: Toxic/Corrosive (e.g., Chlorine, Ammonia).
Grey: Nitrogen, Air,Oxygen.
Green: Argon.
Black: Carbon Dioxide.
Brown: Helium.
422. What are different types of electrode used in Acid-Base, Redox and Precipitation potentiometric titrations?
Acid-Base Titration: pH Glass Electrode
Redox Titration: Inert metallic electrode, typically Platinum (Pt)
Precipitation Titration: A metal electrode corresponding to the precipitate, often a Silver (Ag) electrode for titrations involving silver ions.
423. What is Finger print region in IR?
The “fingerprint region” in IR spectroscopy (Infrared Spectroscopy) refers to the complex area of the spectrum, typically from 400 to 1500 cm⁻¹, containing numerous absorption peaks unique to each compound, much like a human fingerprint.
424. What is polarized light?
Polarized light is light where the electric/magnetic field oscillations are restricted to a single direction.
425. What is Plane polarized light?
Plane polarized light is light where the electromagnetic wave’s vibrations are restricted to a single plane.
426. What are different types of drug dosage forms?
Solid: Tablet, Capsule, Powder, Granules, Suppositories etc.
Liquid: Syrups, Suspension, Drops, Mouthwash etc.
Semi Solids: Ointment, Creams, Gels, Pastes etc.
Other Dosage: Injections, Salines, Inhalers, Transdermal Patches etc.
427. What is leak test ?
A leak test in the pharmaceutical industry is a quality control process to check the integrity of a product’s packaging to ensure it is properly sealed and free from any breaches.
428. What Pareto Chart?
A Pareto chart is a QC tool that displays the 80/20 rule, which suggests that roughly 80% of effects/problems come from 20% of causes.
80% of complaints come from 20% of customers OR 80% of sales come from 20% of clients.
429. What is 5-S in Quality?
5S in quality is a foundational Lean methodology for workplace organization, improving efficiency, productivity, and quality by focusing on five steps: Sort, Set in Order, Shine, Standardize, and Sustain.
430. What is Good Laboratory Practice (GLP)?
Good Laboratory Practice (GLP) principles refers to a set of quality and validity standards for the organisation and management of test facilities and the data they produce.
431. What is Standard Deviation (SD)?
Standard deviation (∂) measures how spread out numbers in a dataset are from the average (mean).
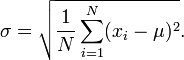
xi is an individual value; μ is the mean/expected value; N is the total number of value.
432. What is MLT test in Microbiology?
A microbial limit test (MLT) is a microbiological test performed on non-sterile products, such as pharmaceuticals, healthcare items, and cosmetics, to check for microbial contamination.
433. What is TAMC?
Total Aerobic Microbial Count (TAMC):Measures the total number of aerobic bacteria.
434. What is TYMC?
Total Combined Yeast and Mold Count (TYMC):Measures the total number of yeast and mold.
435. What is the difference between aerobic and anaerobic bacteria?
Aerobic bacteria require oxygen for survival and growth.
The bacteria that grow in the absence of oxygen are called anaerobic bacteria.
436. What is Pathogen?
A pathogen is an infectious agent that has the potential to cause disease in a host organism.
437. What are names of Pathogens?
1. Escherichia coli
2.Salmonella
3. Pseudomonas aeruginosa
4. Staphylococcus aureus
438. What is BET test in microbiology?
The Bacterial Endotoxin Test (BET) is performed to detect pyrogens (fever-causing substances) in injectable drugs, water, and medical devices, ensuring patient safety by preventing fever, shock, and death from contaminants (endotoxins from Gram-negative bacteria) before they enter the bloodstream or spinal fluid.
439. What is Contact plate and Settle plate method in Microbiology?
In microbiology, settle plates passively collect airborne microbes by exposing open agar dishes to the air for a set time, showing where contaminants fall, while contact plates (also called Rodac plates) press their convex agar surface directly onto surfaces (floors, equipment) to detect microbes on that surface, both methods are key for environmental monitoring (like in cleanrooms).
440. What are the two types of air sampling methods?
The two main types of air sampling methods are Active Sampling (using pumps to draw air through a collection device) and Passive Sampling (relying on natural air diffusion to collect contaminants).
441. What is PM 2.5 and PM 10 in Air Quality Index(AQI)?
PM2.5 and PM10 in the Air Quality Index (AQI) refer to Particulate Matter of different sizes: PM10 are particles under 10 micrometers (like dust, pollen), while PM2.5 are finer, under 2.5 micrometers (like smoke, vehicle exhaust), both measured in micrograms per cubic meter, with smaller PM2.5 posing a greater health risk as they deeply penetrate lungs and enter the bloodstream.
442. What is air flow pattern test in HVAC system?
An HVAC airflow pattern test visually assesses ventilation effectiveness, often using smoke to map air movement (laminar vs. turbulent), identifying stagnant zones or contamination paths, crucial for cleanrooms.
443. What is recovery test in Clean room HVAC system?
a Recovery Test (or Room Recovery Test) checks how fast a controlled space (like a cleanroom or pharma facility) returns to its specified clean conditions (temperature, humidity, particle levels) after a temporary disturbance or contamination event, proving the HVAC system maintains airflow, filtration, and pressure to meet standards like ISO 14644.
444. What is Lux in light?
Lux (lx) is the SI unit for illuminance, measuring the intensity or amount of light falling on a specific surface area.
445. What is lux light requirement in pharma
Lux requirements in pharma vary by area, with general production area needing around 500 lux, sampling/dispensing 300+ lux, labs 500-1500 lux, packaging 300-750 lux, and storage often below 50 lux for sensitive items, while critical visual inspection (especially for vials) can demand extremely high levels like 2000-3750 lux or even 10,000 lux for colored containers to ensure quality and GMP compliance.
446. What are the types forced degradation in method validation?
The main types of forced degradation (stress testing) are Hydrolysis (acid/base), Oxidation, Photolysis (light), and Thermal Degradation (heat/humidity).
Hydrolysis: Testing the drug’s stability in water under acidic (e.g., HCl) and basic (e.g., NaOH) conditions, simulating pH changes.
Oxidation: Exposing the drug to oxidizing agents, commonly hydrogen peroxide (H₂O₂), to check for oxidative damage.
Photolysis: Subjecting the drug to UV and visible light (using specific lamps) to assess light sensitivity, which often leads to opaque packaging.
Thermal Degradation: Applying dry and/or moist heat (elevated temperatures) to see how heat affects the substance, mimicking accidental storage issues.
Humidity Stress: Testing the impact of moisture/humidity, often combined with heat.
447. What are the acceptance criteria for forced degradation?
Degradation of at least 5 % to 30 % should be observed in any of the conditions.
448. What is Schedule-M?
Schedule M is the Indian government’s set of guidelines, under the Drugs and Cosmetics Act, 1940, and 1945, that specifies the requirements for Good Manufacturing Practices (GMP) for pharmaceutical products.
449. What is Schedule-L1?
In Indian pharmacy regulations, Schedule L (now primarily Schedule L1 refers to the Good Laboratory Practices (GLP), outlining mandatory standards for pharmaceutical quality control labs, covering premises, personnel, equipment, testing, quality systems, and data integrity to ensure accurate analysis and reliable product safety.
450. What is Schedule-H?
Schedule H drugs are potent prescription-only medicines listed under the Indian Drugs and Cosmetics Act, requiring a doctor’s prescription for purchase, and must have an “Rx” symbol on their label.
451. What is Solution Stability?
Solution stability proves how long your prepared standard and sample solutions remain accurate and reliable before use. A solution degradation or concentration changes due to factors like temperature, light, or time. By comparing measurements of aged solutions with fresh solution, an acceptable storage period for solution can be set.
452. What is difference between Hold Time and Stability Study?
Hold Time studies check product quality during temporary pauses in manufacturing (e.g., granules waiting for compression), ensuring stability for hours or days, while Stability Studies determine the product’s overall shelf life (months/years) under various conditions (temp, humidity, light) to set expiry dates and storage requirements for the finished, packaged drug.
453. What is Autoclaving in microbiology?
An autoclaving is process in machine that uses high-pressure, saturated steam (like a super-powered pressure cooker) to achieve sterilization, effectively killing all microorganisms, including tough bacterial spores, viruses, and fungi, by exposing them to high temperatures (typically 121°C or 250°F) for a set time (typically 15 to 30 minutes).
454. What is Primary standard?
A primary standard/solution is made from a highly pure, stable, and non-hygroscopic substance that is used to determine the concentration of secondary standard solutions.
455. What is Secondary standard ?
A secondary standards are less pure, less stable, and hygroscopic substance. A solution is prepared and then standardized against a primary standard to find its exact concentration for use in routine analysis.
456. What is expiry period for primary standard solution and secondary standard solution?
The Primary standard solution and secondary standard solution shall be verified periodically and base on results expiry period can be decided.
457. What is freshly prepared Solution?
It is generally means preparation for immediate use or within a very short window (e.g., hours, same day, or within 24 hours), depending on the specific compound.
458. What is shelf life for Volumetric Solutions?
Generally Volumetric Solutions are stable for around 30 days at room/dark conditions.
459. What is Re-standardization frequency for Volumetric solutions?
Re-standardize all volumetric solutions after every 15 days to confirm Molarity/Normality.
460. What is Re-standardization frequency for Unstable Volumetric solutions?
Re-standardize all unstable volumetric solutions before use Or prepare fresh solution and standardize.
461. What is shelf life for Laboratory Reagents/Indicators?
Generally, Laboratory Reagents/Indicators shall be used within 90 days from date of preparation provided solution is stable and it is stored at room/dark conditions.
462. How to store Light Sensitive solutions?
Light sensitive solutions shall be stored in Amber color glass bottle or container.
463. What is Solution Degradation?
Fungal growth, Precipitation, Recrystallization, Turbidity, Color change, or foul odor appears in the solution. Discard solution immediately if any of the above sign observed.
464. How to Determine Shelf Life of Lab solutions?
- Prepare the solution and test its molarity in triplicate.
- Repeat testing at intervals (e.g., every 6 days for 30 days).
- Calculate the % Relative Standard Deviation (%RSD) of the molarity values.
- If %RSD is low (e.g., <1%), the shelf life might be extended.
465. What are the factors which impacts solution stability?
Light, temperature (heat accelerates degradation), air exposure, dirty bottles, caps on unclean surfaces and contamination significantly impacts on solution stability.
466. What is Sedimentation?
Sedimentation is the natural process where solid particles suspended in a fluid (water or air) settle down and accumulate at the bottom due to gravity, forming layers of sediment.
467. What is SUPAC guideline?
SUPAC stands for Scale-Up and Post-Approval Changes. It refers to a set of U.S. Food and Drug Administration (FDA) guidance documents that outline the necessary chemistry, manufacturing, and controls (CMC) documentation and testing requirements for changes made to an approved new drug application (NDA) or abbreviated new drug application (ANDA).
468. What does level 1, 2, and 3 mean on SUPAC?
Level I changes are minor, Level II are moderate, and Level III are major.
469. What is a SUPAC Level-3 change?
Level 3 changes are those that are likely to have significant impact on formulation quality and performance of drug product.
470. What is a SUPAC Level-2 change?
Level 2 changes are those changes that have a moderate impact on product quality and may require additional documentation or stability testing.
471. What is type B cleaning in pharma?
Type B cleaning in pharma is a major cleaning procedure for equipment and areas performed when switching between different products, after a major maintenance event, or when cleaning hold times are exceeded.
472. Why 900 ml media volume in dissolution?
The 900mL volume is used in dissolution testing to ensure “sink conditions,” which means there is enough dissolution media to completely dissolve the drug at any given time, preventing the drug concentration from reaching saturation. This large volume ensures that the rate of drug release is not limited by the solubility of the drug, leading to a more accurate and reproducible assessment of the product’s performance.
473. Why are only 6 tablets used in dissolution?
In dissolution testing, n=6 is the typical amount of samples to test to make decisions. This n=6 comes from statistics and is where we have a 95% confidence interval in the data.
474. What is CIP and SIP in pharma?
CIP (Clean-in-Place) and SIP (Sterilize-in-Place) are automated systems that clean and sterilize equipment like tanks and pipes without taking them apart, crucial for GMP compliance. CIP uses water, heat, and chemicals (acids/alkalis) to remove residues, while SIP follows to kill microorganisms using high-temperature steam (e.g., 121°C). CIP achieves ‘visually clean’ status, while SIP ensures sterility for aseptic processes.
475. What is the difference between Uncoated, Film Coated and Enteric coated tablets?
Uncoated Tablets: These are simple compressed tablets made of active ingredients and excipients (inactive substances) without any additional outer layer. Designed to rapid release in stomach.
Coated Tablets: These tablets have an outer layer made of various materials like sugar or a thin polymer film. Designed to mask unpleasant taste or odor, make the tablet easier to swallow, protect the drug from environmental factors (moisture, light), and improve its appearance.
Enteric coated Tablets: A specific type of coated tablet that has a polymer barrier designed to be resistant to the acidic environment of the stomach (pH-sensitive polymers). Ensures the medication is released in the intended site of action, such as the intestines.
476. What is the difference between Aqueous and Non aqueous titration?
The Aqueous titration uses water, while Non-aqueous titration uses an organic solvent.
477. What is the principle of LAF?
The principle of laminar air flow (LAF) involves a continuous, unidirectional flow of filtered air to create a sterile environment.
478. Why are 0.3 micron HEPA filters used in LAF?
HEPA filter captures 99.97% of particles that are 0.3 microns or larger, including dust, bacteria, etc. Placed after the pre-filter, the HEPA filter ensures that only clean, uncontaminated air enters the work area.
A Laminar Air Flow hood (LAF) protects the product/sample from airborne particles providing a sterile, unidirectional airflow (vertical or horizontal) from HEPA-filtered air, ideal for non-hazardous work ; while a Biosafety Cabinet (BSC) protects the product, user, and environment by using inward airflow and HEPA filtration for both incoming and outgoing air, essential for handling infectious or hazardous biological materials.
480. What is the difference between LAF and RLAF?
LAF (Laminar Air Flow) pushes clean air down (or horizontally) to protect the product/sample from room contamination, like a sterile blanket. RLAF (Reverse Laminar Air Flow) pulls air inward from the operator’s side towards the back/bottom, creating a contaminated-to-clean flow to protect both the operator and the product from hazardous powders or aerosols during handling (like dispensing/sampling).
481. What is Air Shower?
Air shower is a self-contained chamber at cleanroom entrances/exits that blasts high-velocity, HEPA-filtered air to remove dust, germs, and particles from personnel and equipment, preventing contamination by creating a barrier and ensuring ultra-clean environments for drug manufacturing, biotech, and labs.
482. What is the role of epoxy flooring in pharma?
Epoxy flooring plays a crucial role by providing seamless, durable, and hygienic surfaces that meet strict GMP regulations, preventing microbial growth, and ensuring easy cleaning for sterile environments in processing, labs, and packaging areas. Its smooth, non-porous nature eliminates joints where bacteria & debris can hide and making it easy to clean and sanitize.
483. What is Signal to Noise Ratio in HPLC?
Signal (H): The height of the analyte peak.
Noise (h): The random fluctuations in the baseline, often measured over a defined noise-free window
S/N = 2H/h
484. What is the Signal to Noise ratio criteria for LOD & LOQ?
Limit of Detection (LOD): S/N=3
Limit of Quantification (LOQ): S/N=10
485. What is Pharmacovigilance?
Pharmacovigilance is the science and activities that involve detecting, assessing, understanding, and preventing adverse effects of drug and related problems. It continuously monitors the safety of medicines post market in real-world use.
486. What is Temperature Mapping?
Temperature mapping is the systematic process of using sensors to monitor and document temperature variations within storage areas (warehouses, fridges, cold rooms) to ensure all spots meet required ranges, identifying hot/cold spots, validating equipment, and guaranteeing the safety, quality, and efficacy of temperature-sensitive medicines, all while adhering to strict regulatory standards like USP and WHO guidelines.
487. What is Hot and Cold spot in temperature mapping?
Hot spots are the areas with the highest recorded temperatures, while cold spots are the areas with the lowest temperatures.
488. What is a cold spot in an Autoclave?
A “cold spot” in an autoclave is the area where the temperature rises the slowest, and consequently, is the last to reach the required sterilization temperature.
489. Why 121°C temperature used in Autoclave?
Autoclave uses 121°C because it’s a universally effective temperature for killing heat-resistant bacterial spores (like Bacillus and Clostridium) using moist heat from pressurized steam, while also balancing effective sterilization with protecting heat-sensitive materials like plastics and rubber, achieving this at a standard pressure of about 15 psi for 15-20 minutes.
490. What is the procedure for handling of poisonous chemicals in laboratory?
Handling poisonous lab chemicals involves strict adherence to PPE (gloves, goggles, lab coats), working under a fume hood for volatiles, proper storage in labeled, locked cabinets, meticulous inventory, reviewing the chemical’s Safety Data Sheet (SDS) for specific risks and emergency steps, including using spill kits and having emergency contacts ready for quick response.
491. Why Analytical Method validation required ?
To ensure methods for testing of drugs produce reliable, accurate, and consistent result which is guaranteeing product quality, patient safety, and regulatory compliance (FDA, EMA).
492. Why is it necessary to validate the Analyst?
The analyst needs to be validated for his ability to generate authentic and reliable testing results in laboratory. It gives assurance that analyst is well qualified & trained to perform testing to give reliable,consistent and accurate results.
493. What is the pH of purified water as per USP?
As per USP Pharmacopeia, purified water must have a pH between 5.0 and 7.0
494. What is the conductivity of purified water as per USP?
Conductivity should be less than 1.3𝜇𝑆/𝑐𝑚 at 25°𝐶 according to USP.
495. What causes high conductivity in purified water?
when water contains dissolved ions (e.g., sodium chloride, calcium, magnesium, etc.) and dissolve carbon dioxide (𝐶𝑂2) the conductivity increases.
496. How to reduce high conductivity in water?
To reduce water conductivity, need to remove dissolved ions (salts/minerals) using methods like Reverse Osmosis (RO), Distillation, or Ion Exchange, which physically filter or swap ions out, making water purer and less conductive. Removing dissolved CO2 by degassing.
497. What is ETP plant in pharma?
An ETP plant (Effluent Treatment Plant) is a crucial wastewater treatment system that purifies industrial wastewater from drug manufacturing, removing harmful chemicals, APIs, heavy metals, and solids using physical, chemical, and biological processes before discharge or reuse, ensuring environmental compliance and sustainability.
498. What COD & BOD in water?
BOD (Biochemical Oxygen Demand): The amount of dissolved oxygen (DO) consumed by microorganisms (bacteria) to break down biodegradable organic material in water.
COD (Chemical Oxygen Demand): The amount of oxygen needed to chemically oxidize all organic and some inorganic matter in water.
499. What is TSE/BSE Certificate?
A TSE/BSE certificate is a document that verifies a product, often made from animal by-products, is free from dangerous animal-borne diseases or has a minimized risk of Transmissible Spongiform Encephalopathy (TSE), such as Bovine Spongiform Encephalopathy (BSE), also known as “mad cow disease” (fatal brain diseases caused by infectious proteins called prions)
500. What is the maximum temperature use for volumetric glassware drying?
For routine lab glassware drying, 60°C is ideal. Prolonged exposure to above 60°C can cause microfractures and stress to the glassware which can disturb calibration volume of glassware.