
Topic:– Process Validation
Definition:
“process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product .”
Guidelines – 1
–FDA/ICH, (CDER and CBER),
Q7-Good Manufacturing Practice for Active Pharmaceutical Ingredients, guidance for industry, August 2001.
–FDA/ICH, (CDER and CBER),
Q8(R2)-Pharmaceutical Development, guidance for industry, November 2009.
–FDA/ICH, (CDER and CBER),
Q9 -Quality Risk Management, guidance for industry, June 2006.
–FDA/ICH (CDER and CBER)
Q10- Pharmaceutical Quality System, guidance for industry, April 2009.
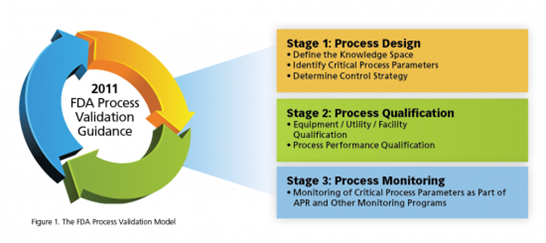
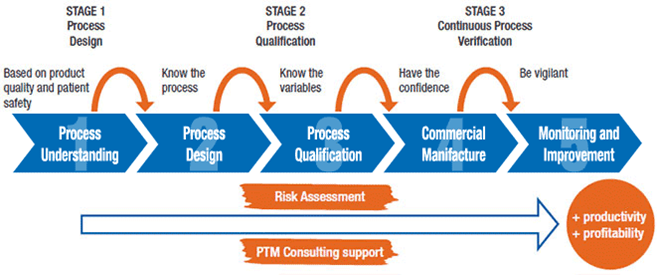
Process validation involves a series of activities taking place over the lifecycle of the product and process. This guidance describes process validation activities in three stages.
Stage 1 – Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities.
Stage 2 – Process Qualification: During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.
Stage 3 – Continued Process Verification: Ongoing assurance is gained during routine production that the process remains in a state of control.
A successful validation program depends upon information and knowledge from product and process development. This knowledge and understanding is the basis for establishing an approach to control of the manufacturing process that results in products with the desired quality attributes. Manufacturers should:
–Understand the sources of variation.
–Detect the presence and degree of variation.
–Understand the impact of variation on the process and ultimately on product attributes.
–Control the variation in a manner commensurate with the risk it represents to the process and product.
FDA regulations describing current good manufacturing practice (CGMP) for finished pharmaceuticals are provided in 21 CFR parts 210 and 211.
The CGMP regulations require that manufacturing processes be designed and controlled to assure that in-process materials and the finished product meet predetermined quality requirements and do so consistently and reliably. Process validation is required, in both general and specific terms, by the CGMP regulations in parts 210 and 211. The foundation for process validation is provided in § 211.100(a), which states that “[t]here shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess…” (emphasis added). This regulation requires manufacturers to design a process, including operations and controls, which results in a product meeting these attributes.
Many products are single-source or involve complicated manufacturing processes. Homogeneity within a batch and consistency between batches are goals of process validation activities. Validation offers assurance that a process is reasonably protected against sources of variability that could affect production output, cause supply problems, and negatively affect public health.
Stage 1 ― Process Design
Process design is the activity of defining the commercial manufacturing process that will be reflected in planned master production and control records. The goal of this stage is to design a process suitable for routine commercial manufacturing that can consistently deliver a product that meets its quality attributes.
Product development activities provide key inputs to the process design stage, such as the intended dosage form, the quality attributes, and a general manufacturing pathway. Process information available from product development activities can be leveraged in the process design stage. The functionality and limitations of commercial manufacturing equipment should be considered in the process design, as well as predicted contributions to variability posed by different component lots, production operators, environmental conditions, and measurement systems in the production setting. However, the full spectrum of input variability typical of commercial production is not generally known at this stage. Laboratory or pilot-scale models designed to be representative of the commercial process can be used to estimate variability.
Designing an efficient process with an effective process control approach is dependent on the process knowledge and understanding obtained. Design of Experiment (DOE) studies can help develop process knowledge by revealing relationships, including multivariate interactions, between the variable inputs (e.g., component characteristics 13 or process parameters) and the resulting outputs (e.g., in-process material, intermediates, or the final product). Risk analysis tools can be used to screen potential variables for DOE studies to minimize the total number of experiments conducted while maximizing knowledge gained. The results of DOE studies can provide justification for establishing ranges of incoming component quality, equipment parameters, and in-process material quality attributes. FDA does not generally expect manufacturers to develop and test the process until it fails.
Stage 2 ― Process Qualification
During the process qualification (PQ) stage of process validation, the process design is evaluated to determine if it is capable of reproducible commercial manufacture. This stage has two elements: (1) design of the facility and qualification of the equipment and utilities and (2) process performance qualification (PPQ). During Stage 2, CGMP-compliant procedures must be followed. Successful completion of Stage 2 is necessary before commercial distribution.15 Products manufactured during this stage, if acceptable, can be released for distribution.
Qualification of utilities and equipment generally includes the following activities:
Selecting utilities and equipment construction materials, operating principles, and performance characteristics based on whether they are appropriate for their specific uses.
Verifying that utility systems and equipment are built and installed in compliance with the design specifications (e.g., built as designed with proper materials, capacity, and functions, and properly connected and calibrated).
Verifying that utility systems and equipment operate in accordance with the process requirements in all anticipated operating ranges. This should include challenging the equipment or system functions while under load comparable to that expected during routine production. It should also include the performance of interventions, stoppage, and start-up as is expected during routine production. Operating ranges should be shown capable of being held as long as would be necessary during routine production.
The process performance qualification (PPQ) is the second element of Stage 2, process qualification. The PPQ combines the actual facility, utilities, equipment (each now qualified), and the trained personnel with the commercial manufacturing process, control procedures, and components to produce commercial batches. A successful PPQ will confirm the process design and demonstrate that the commercial manufacturing process performs as expected. The approach to PPQ should be based on sound science and the manufacturer’s overall level of product and process understanding and demonstrable control. The cumulative data from all relevant studies (e.g., designed experiments; laboratory, pilot, and commercial batches) should be used to establish the manufacturing conditions in the PPQ.
In most cases, PPQ will have a higher level of sampling, additional testing, and greater scrutiny of process performance than would be typical of routine commercial production. The level of monitoring and testing should be sufficient to confirm uniform product quality throughout the batch. The increased level of scrutiny, testing, and sampling should continue through the process verification stage as appropriate, to establish levels and frequency of routine sampling and monitoring for the particular product and process. Considerations for the duration of the heightened sampling and monitoring period could include, but are not limited to, volume of production, process complexity, level of process understanding, and experience with similar products and processes.
Stage 3 ― Continued Process Verification
The goal of the third validation stage is continual assurance that the process remains in a state of control (the validated state) during commercial manufacture. A system or systems for detecting unplanned departures from the process as designed is essential to accomplish this goal. Adherence to the CGMP requirements, specifically, the collection and evaluation of information and data about the performance of the process, will allow detection of undesired process variability. Evaluating the performance of the process identifies problems and determines whether action must be taken to correct, anticipate, and prevent problems so that the process remains in control (§ 211.180(e)).
An ongoing program to collect and analyze product and process data that relate to product quality must be established (§ 211.180(e)). The data collected should include relevant process trends and quality of incoming materials or components, in-process material, and finished products. The data should be statistically trended and reviewed by trained personnel. The information collected should verify that the quality attributes are being appropriately controlled throughout the process.
Data gathered during this stage might suggest ways to improve and/or optimize the process by altering some aspect of the process or product, such as the operating conditions (ranges and set-points), process controls, component, or in-process material characteristics. A description of the planned change, a well- justified rationale for the change, an implementation plan, and quality unit approval before implementation must be documented (§ 211.100). Depending on how the proposed change might affect product quality, additional process design and process qualification activities could be warranted.
Maintenance of the facility, utilities, and equipment is another important aspect of ensuring that a process remains in control. Once established, qualification status must be maintained through routine monitoring, maintenance, and calibration procedures and schedules (21 CFR part 211, subparts C and D).
The equipment and facility qualification data should be assessed periodically to determine whether re-qualification should be performed and the extent of that re-qualification. Maintenance and calibration frequency should be adjusted based on feedback from these activities.
Disclaimer:-This presentation is solely prepared for training, sharing knowledge and information purpose only collected from various guidelines and literature. The information presented here is only for reference purpose.