
Topic– Method Validation
Definition:
Validation is a process of establishing documentary evidence demonstrating that a procedure, process or activity carried out in the production or testing maintains the desired level of compliance at all stages.
Analytical method validation is a process of documenting/proving that an analytical method provides analytical data acceptable for the intended use.
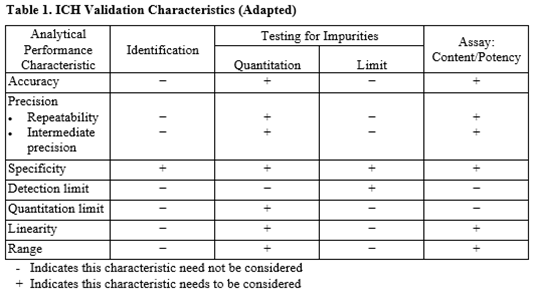
Guidelines – 1
–-Q2 (R1)-Validation of analytical procedures : Text and Methodology.
–USP <1225>Validation of compendial procedures
–USP <1226>Verification of compendial procedures
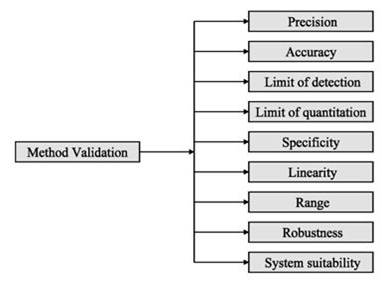
* Specificity
* Force Degradation (If Applicable)
* Precision
-
Repeatability
-
Intermediate precision
-
Reproducibility
*Linearity and Range
*Accuracy
*Robustness
*Limit of detection (LOD) and Limit of Quantitation (LOQ
*Solution stability
Specificity:
Ability to asses unequivocally the analyte in the presence of components which may be expected to be present (Impurities, degradants)
For assay and impurity methods the samples shall contain materials which are potentially present during routine analysis and may interfere with the result, e.g. analysis sample containing potential interferences, e.g. impurities, Excipients.
Sample shall prepared by spiking drug substance / drug product with potentially interfering material. The acceptance criteria shall be; No interference observed for response due to analyte or impurities or interest and no peaks interfering with analyte peak or the peaks due to the impurities of interest observed. Peak purity shall not be less than 0.995.
For the impurities test, the determination shall be established by spiking drug substances or drug product with appropriate levels of impurities and demonstration the separation of these impurities individually or from other components in the sample matrix.
Forced degradation:-
Forced degradation shall be done in presence of excipients, firstly performed during the pre-formulation stage to assist in the selection of the most formidable compounds and excipients. This shall lead to the development of more suitable formulation, packaging and change in storage and manufacturing conditions as the optimal formulation is defined to be used. Forced degradation studies are also in order to demonstrate specificity during the validation of stability indicating methods.
These studies are usually performed at conditions exceeding that of accelerated storage condition.
Forced degradation studies shall provide information to degradation pathway and degradation products that could form during storage of the drug product.
The extent of targeted degradation shall be approximately anywhere from 5% to 20%. In some cases the degradation can be difficult to achieve 5%, in such cases statement shall be made in the validation report that the product quality / efficacy shall not be affected for such particular stress condition. The assessment of peak purity using diode array shall be employed.
Precision:
Precision is the closeness of agreement (degree of scatter) between a series of measurements obtained from multiple sampling of the same homogeneous sample. There are following aspects to achieve for precision.
-
Repeatability:
Repeatability is a measure of precision under the same operating conditions over a short interval of time that is, under normal operating conditions of analytical method with the same equipment.
Repeatability to be accessed using a minimum of nine determinations covering the specified range for the procedure (e.g. three concentrations / three replicates as in the accuracy experiment) of using a minimum six determination at 100% of the test concentration. Standard deviation, relative standard deviation and confidence interval to be reported. The assay value of independent analysis of sample preparation to final test result. For the related substances, residual solvents precision shall be established at 100% specification level of know impurities / organic solvents and also to be established at the limit of quantification level.
b. Intermediate precision:
Intermediate precision is defined as the variation within the same laboratory. The extent to which intermediate precision needs to be established depends on the circumstances under which the procedure is intended to be used. Typical parameters that are investigated include day to day variation, analyst variation and equipment variation. Depending upon the extent of the study, the use of experimental design is encouraged.
Experimental design shall be minimize the number of experiments that need to be performed. It is from doing intermediate precision when reproducibility is proven. It shall be expected that the intermediate precision shall show variability that is in same range or less than repeatability variation. The standard deviation, relative standard deviation (coefficient variation) and confidence interval to be established of data.
c. Reproducibility:
Reproducibility measures the precision between laboratories. This parameter shall be considered in the standardization of an analytical method (e.g. inclusion of procedures in pharmacopoeias and method transfer between different laboratories).
To validate this characteristic, similar study to be performed at different laboratories using the same homogeneous sample lot and the same experimental design. In case of method transfer between two laboratories, different approaches may be taken to achieve the successful transfer of the procedure. The approach is method transfer from the originating laboratory to the receiving laboratory. The originating laboratory is defined as the laboratory that has developed and validated analytical method. The receiving laboratory is defined as laboratory to which the analytical method to be transferred and that will participate in the method transfer studies. In the method transfer, it is recommended that protocol be initiated with details of the experiments to be performed and acceptance criteria (in term of the difference between the means of the two laboratories) for passing the method transfer.
Linearity:-
Linearity of an analytical procedure as the ability (within the given range) to obtain test results of variable data (e.g. absorbance and area under the curve) which are directly proportional to the concentration (amount of analyte) in the sample. The data variables that shall be used for quantitation of the analyte are the peak area or ration of peak area of analyte to the internal standard peak. The working sample concentration and samples tested for accuracy shall be in the linear range.
There are two general approaches for determining the linearity of the method.
The first approach is to weigh different amounts of standard directly to prepare linearity solutions at different concentrations. However, it is not suitable to prepare solution at very low concentration, as the weighing error shall be relatively high.
Another approach is to prepare a stock solution of high concentration. Linearity is then demonstrated directly by dilution of standard stock solution. This is more popular and the recommended approach.
Linearity is best evaluated by visual inspection of a plot of signals as a function of analyte concentration. Subsequently, the variable data are generally used to calculate a regression line by the least square method. At least five concentration levels shall be used.
Under normal circumstances, linearity shall be with coefficient of determination (r2) of 0.995 the slope, residual sum of squares, and y- intercept shall also be reported.
The slope of the regression line shall provide an idea of the sensitivity of the regression. The y intercept shall provide an estimate of the variability of the method. The ratios percent of the y intercept with the variable data at nominal concentration are used to estimate the method variability.
For determination of assay of drug substances of a drug product, the usual range of linearity shall be ±20% of the target or nominal concentration. For the determination of content uniformity, it shall be ±30% of the target of nominal concentration. For determination of related substances or residual solvents it shall be from the reporting level to 20% ahead of the target or nominal concentration. For dissolution testing: ±20% over the specified range.
Limit of Detection (LOD):-
It is the lowest amount of analyte in a sample that shall be detected but not necessarily quantitated under the stated experimental conditions.
The detection shall usually expressed as the concentration of the analyte in the sample, e.g. percentage, parts per million (ppm) or parts per billion (ppb).
There are several approaches to establish the limit of detection. One approach to establish detection limit shall be determined by the analysis of a series of sample with known concentrations and establishing the minimum level at which analyte shall be reliably detected. For instrumental procedures that exhibit background noise, it shall compare measured signals from samples with known concentrations of analyte with those of the blank samples. The minimum concentration at which the analyte shall reliably be detected, shall establish using an acceptable signal to noise ratio of 2:1 or 3:1.
Presentation of relevant chromatograms shall be sufficient for justification of detection limit.
Limit of Quantitation (LOQ):-
Quantitation limit is determined by analysis of samples with known concentrations of analyte and by establishing the minimum level at which the analyte shall be quantitated with acceptable accuracy and precision.
signal to noise ratio by comparing measured signals from samples with known low concentration at which the analyte shall be reliable quantified at the signal to noise ration 10:1.
Accuracy:-
Accuracy of an analytical procedure as the closeness of agreement between the values that are accepted either as conventional true values or an accepted references value and the value found. For the drug substances accuracy is defined by the application of the analytical procedure to an analyte of known purity (e.g. reference standard). For the drug product, accuracy shall be determined by application of the analytical procedure to synthetic mixtures of the drug product components to which amounts of analyte have been added within the range of the procedure. It is recommended to assess minimum of nine determinations over a minimum three concentration levels covering the specified range (e.g. three concentration / three replicates).
Accuracy shall be reported as percent recovery by assay (using the proposed analytical procedure) of known added amount of analyte in the sample of as the difference between the mean and the accepted true value together with the confidence intervals. The range for the accuracy limit shall be within the linear range. Typically accuracy of the recovery of the drug substance is expected to be about 99 – 101%.
Typical accuracy of drug product is expected to be 98 – 102%. For the dissolution of drug product accuracy of recovery is expected to be 98 – 102%. Values of accuracy of recovery data beyond this range need to be investigated as appropriate.
For the related substances / residual solvents recovery shall be performed from the quantitation limit by spiking the known concentration of known impurities / residual solvents in analyte and the accuracy of recovery can be up to 85 – 115%.
For Cleaning validation recovery shall be performed from the quantitation limit to minimum 120% by spiking the known concentration of known analyte and the accuracy of recovery should not be less than 70%.
Robustness:-
Robustness of an analytical procedure is a measure of the analytical method to remain unaffected by small but deliberate variations in method parameter and provides an indication of its reliability during normal usage.
Common method parameters that can affect the analytical procedure shall be considered based on the analytical technique;
Sample preparation
Extraction time
High performance liquid chromatography (HPLC) conditions;
Mobile phase composition (pH ±0.05, percent organic ± 2% absolute) Column used (equivalent columns, lots and / or suppliers) Temperature (± 5º C)
Flow rate (± 10 %)
Gas chromatography (GC) conditions
Column used (equivalent columns, lots and / or suppliers) Temperature (± 10 %)
Flow rate (± 10 %)
When the results are affected by some critical experimental parameters, a precautionary statement shall be included in the analytical procedure to ensure that this parameter is tightly controlled between experiments. For example: in case percent ion pairing of mobile phase affects the results significantly, the analytical procedure shall explicitly be written with a precautionary statement for aqueous component.
Solution stability:-
Stability of Standard and Sample Solution shall be performed at various time intervals and at room temperature.
Acceptance criteria shall be as follows :
For assay and dissolution, the relative standard deviation for peak area as obtained from standard solution and test solution shall not be more than 2.0%.
For related substance and residual solvents, the relative standard deviation for peak area as obtained from standard solution and test solution shall not be more than 10.0 % at various time intervals.
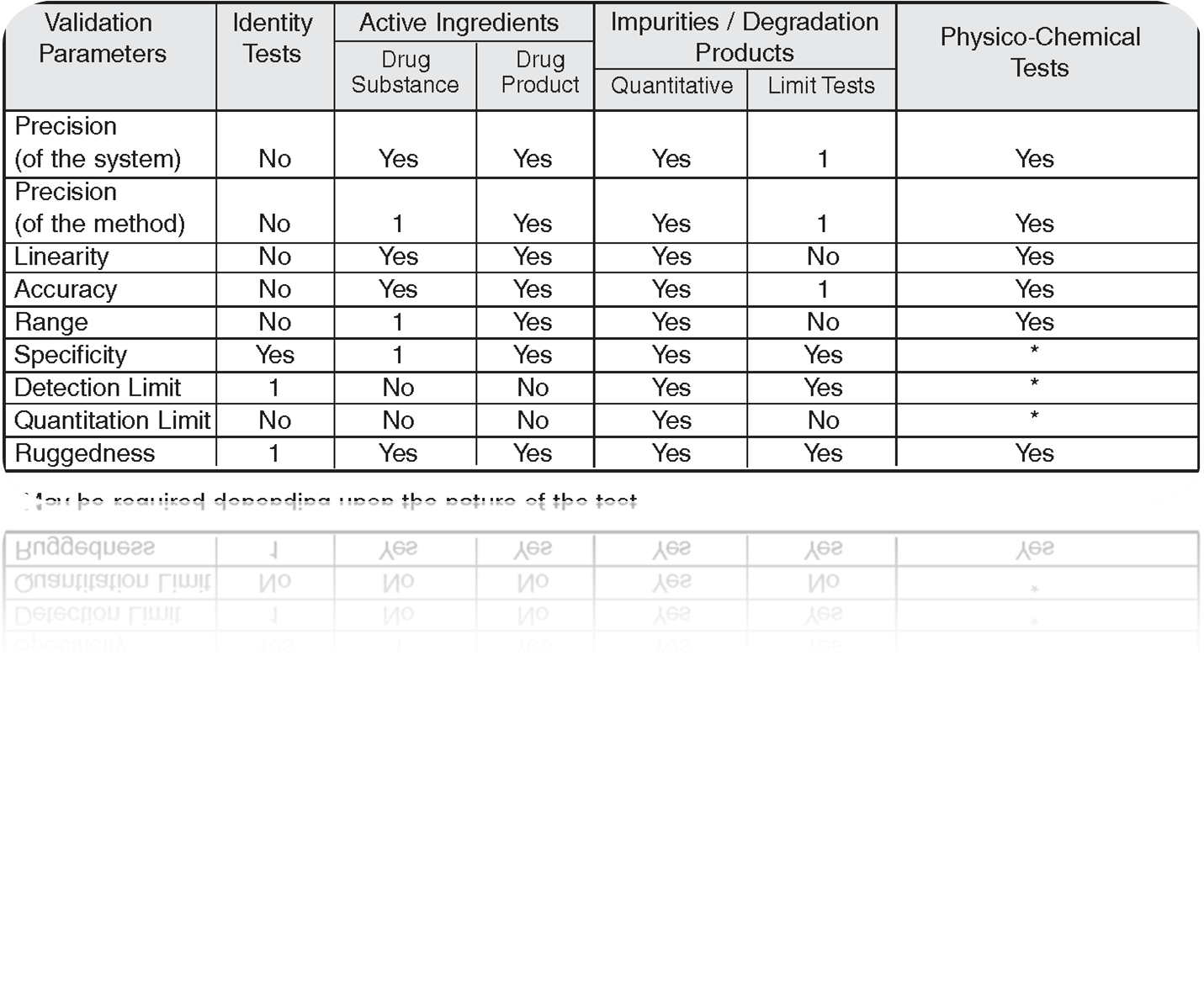
Method Verification:-
Verification consists of assessing selected analytical validation characteristics described earlier to generate appropriate relevant data rather than repeating the validation process for commercial products. The method verification shall be done for the pharmacopeial methods (compendial methods) such as titrations, chromatographic procedures (related compounds, assay, dissolution and limit tests), and spectroscopic tests.
However, general tests (water, heavy metals, residue on ignition) do not typically require verification.
Range:-
The range of analytical procedure is the interval between the upper and lower concentration of analyte in the sample for which it has been demonstrated that the analytical procedure shall a suitable level of precision, accuracy, and linearity. The range shall normally expressed in the same units as test results (e.g. percent, parts per million) obtained by the analytical procedure.
Method Revalidation:-
The analytical method shall be revalidation under following circumstances; however these circumstances are not limited;
In case new impurity found that makes deficient in its specificity, this method needs to be modified and revalidated.
Changes in the excipients composition can change the product impurity profile.
Changes in equipment or supplier of critical supplies of the API (Active Pharmaceutical Ingredients) or final drug product shall have the potential to change their degradation profile and shall require the method to be redeveloped and revalidated.
Disclaimer:-This presentation is solely prepared for training, sharing knowledge and information purpose only collected from various guidelines and literature. The information presented here is only for reference purpose.